Transfusion
Medicine
Handbook
Third Edition,
2016
A Guide to the Clinical Use
of Blood Components,
Blood Products and Blood
Transfusion Procedures in
New Zealand
NEW ZEALAND BLOOD SERVICE
New Zealand Blood Service (NZBS) was formed in 1998 integrating all hospital-based
transfusion services into a single national organisation. Since 2001, NZBS has managed
the recruitment of blood donors, and the collection, processing and accreditation of
all donated blood. This ensures that the demand for blood components used in the
treatment of patients in all public and private hospitals is met.
As well as the collection of blood from donors at many fixed and mobile sites throughout
the country, a wide range of activities are carried out at the main centres. These include
recruitment of donors for high titre immune plasma (anti-D, tetanus, hepatitis B and
zoster immunoglobulin), recruitment of apheresis donors predominantly for plasma
and platelets but also granulocytes, and collection of haemopoietic progenitor cells.
NZBS also carries out therapeutic plasma exchange, therapeutic venesection and
collection of autologous blood.
Other functions of NZBS include specialist immunohaematology, tissue typing
for transplantation, skin and bone banking, production of serum eye drops and
cryogenic storage of blood components. The national stock of rare blood, national
immunohaematology Reference Laboratory, national Tissue Typing Laboratory and the
New Zealand Bone Marrow Donor Registry are all located at the NZBS Auckland centre.
NZBS is responsible for the collection and coordination of plasma supply to CSL
Behring in Melbourne where New Zealand plasma is fractionated into plasma products,
which NZBS then distributes to hospitals and health professionals.
NZBS maintains close liaison with public and private hospitals, general practitioners
and midwives. Hospital transfusion committees operate in most major hospitals and
NZBS is involved with these in an advisory capacity.
NZBS manages the blood banks in six of the country’s major hospitals, with the
remainder being managed by the District Health Boards (DHBs) or local community
laboratory providers. The blood banks carry out various activities including the final
compatibility checking and issuing of blood components and plasma products for
transfusion.
A national computer system, eProgesa, links NZBS and all the main hospital blood
banks. eProgesa manages the whole transfusion process with full traceability of each
individual blood component from donation to its final fate. Information from eProgesa
allows NZBS to monitor blood component usage and, together with the DHBs,
actively manage demand.
There are many elements to the transfusion process that need to be managed
effectively to ensure blood components are used appropriately, and the relationship
between NZBS and the DHBs is a key part of this. NZBS undertakes audits of clinical
transfusion practice and blood use, monitors transfusion-related adverse events
through a national Haemovigilance programme and provides a wide range of clinical,
nursing and technical oversight and support.
Transfusion Medicine Handbook 3rd Edition
Page 1
CONTENTS
PAGE
1. INTRODUCTION
12
1.1 Audience
12
1.2 Evidence
12
1.3 Clinical practice guidelines
12
1.4 Haemovigilance
13
2. COLLECTION, TESTING AND PROCESSING OF BLOOD DONATIONS 15
2.1 Blood donors
15
2.2 Donor selection criteria
15
2.3 Self-sufficiency and the volunteer status of donors
16
2.4 Informed consent for donation
16
2.5 Apheresis donation
16
2.6 Directed and selected donation
17
2.7 Haemochromatosis
17
2.8 Cord blood donation
17
2.9 Testing of donor blood
18
2.10 Leucodepletion
18
2.11 Processing of collected blood to components
19
2.12 Processing of collected blood to fractionated products
19
2.13 Blood components and fractionated products as medicines
20
3. GUIDE TO GOOD TRANSFUSION PRACTICE
21
3.1 Clinical governance
21
3.2 Prescribing blood components and fractionated products
22
3.3 Informed consent to receive a transfusion
23
3.4 Requesting blood components and fractionated products
23
3.5 Blood stock management: the maximum blood order schedule
24
3.6 Collecting blood samples for pretransfusion testing
24
3.7 Pretransfusion testing
25
3.8 Patients with a positive antibody screen
26
3.9 Sample validity (‘72-hour rule’)
26
3.10 Provision of red cells in an emergency
27
3.11 Removal from storage and time limits for infusion
28
3.12 Administration and observation of transfusion
29
3.13 Rate of infusion and precautions
29
3.14 Infusion pumps
30
3.15 Blood administration sets and filters
30
3.16 Warming of blood products
30
3.17 Compatible intravenous solutions
31
3.18 Adding medication to blood components
31
Page 2
Transfusion Medicine Handbook 3rd Edition
3.19 Documentation of transfusion
31
3.20 Local systems and procedures
32
3.21 Reporting of adverse events
33
4. BLOOD COMPONENTS
34
4.1 ABO blood groups and antibodies
34
4.2 Avoiding abo incompatible transfusions
34
4.3 RhD antigen
35
4.4 Other blood group systems
36
4.5 Cytomegalovirus (CMV)
36
4.6 Irradiation
36
4.7 Blood components available from NZBS
39
4.8 Red blood components
46
4.9 Platelet components
48
4.10 Granulocyte components
51
4.11 Plasma components
52
4.12 Fresh frozen plasma
53
4.13 Cryosupernatant apheresis - high fibrinogen
55
4.14 Cryosupernatant plasma
56
5. FRACTIONATED PRODUCTS
57
5.1 Coagulation factors
58
5.1.1 Biostate® (Factor VIII)
58
5.1.2 MonoFIX®-VF (Factor IX)
60
5.1.3 Prothrombinex®-VF (Factors II, IX and X)
62
5.1.4 FEIBA NF® (Factor VIII inhibitor bypassing fraction)
64
5.1.5 RiaSTAP® (Fibrinogen)
67
5.1.6 Fibrogammin® P (Factor XIII)
68
5.2 Natural inhibitors of coagulation
70
5.2.1 Thrombotrol®-VF (Antithrombin III)
70
5.2.2 Ceprotin (Protein C)
70
5.3 Albumin solutions
71
5.3.1 Albumex® 4 (Human albumin 4%)
71
5.3.2 Albumex® 20 (Human albumin 20%)
73
5.4 Immunoglobulin preparations
76
5.4.1 Normal immunoglobulin-VF
77
5.4.2 Hepatitis B immunoglobulin-VF
80
5.4.3 HyperHEP™ S/D
81
5.4.4 Tetanus immunoglobulin-VF
82
5.4.5 Zoster immunoglobulin-VF
84
5.4.6 Berirab® P (Rabies immunoglobulin)
86
5.4.7 Rh(D) immunoglobulin-VF (Anti-D immunoglobulin)
86
5.4.8 Rhophylac® (Anti-D immunoglobulin)
89
5.4.9 Intragam® P (Normal immunoglobulin, intravenous, IVIg)
90
5.4.10 Privigen® (Normal immunoglobulin, intravenous, IVIg)
93
Transfusion Medicine Handbook 3rd Edition
Page 3
5.4.11 Evogam® (Normal immunoglobulin, subcutaneous, SCIg)
98
5.5 Other products
100
5.5.1 Berinert® P (C1-esterase inhibitor)
100
5.5.2 Products from Australian Red Cross Blood Service (ARCBS) 100
6. SPECIAL CIRCUMSTANCES
101
6.1 Management of acute blood loss
101
6.2 Massive transfusion protocol (MTP)
106
6.3 Complications of acute blood loss associated with large volume
transfusions 108
6.4 Avoidable haemostatic problems in elective surgery
109
6.4.1 Warfarin
109
6.4.2 Non-vitamin K-dependent oral anticoagulants (NOAC)
110
6.4.3 Aspirin
111
6.4.4 Non-steroidal anti-inflammatory drugs (NSAID)
111
6.4.5 P2Y adenosine diphosphate (ADP) receptor inhibitors
111
12
6.4.6 Platelet glycoprotein IIb (GPIIb) and IIIa (GPIIIa) inhibitors
112
6.5 Oral anticoagulant induced bleeding or overdose
113
6.5.1 Warfarin
113
6.5.2 Non-vitamin K-dependent oral anticoagulants (NOAC)
115
6.6 Thrombolytic therapy
117
6.7 Disseminated intravascular coagulation (DIC)
117
6.8 Cardiopulmonary bypass
117
6.9 Haemolytic disease of the fetus and newborn (HDFN)
118
6.10 Intrauterine transfusion (IUT)
123
6.11 Transfusion of the newborn
124
6.12 Neonatal autoimmune thrombocytopenia
126
6.13 Fetal and neonatal alloimmune thrombocytopenia (FNAIT)
127
6.14 Individuals refusing blood transfusion
128
7. ADVERSE EFFECTS OF TRANSFUSION
129
7.1 Overview
129
7.2 Reporting adverse reactions or events
129
7.3 Guidelines for the management of adverse transfusion reactions
131
7.4 Febrile non-haemolytic transfusion reaction
144
7.5 Allergic & anaphylactic transfusion reaction
144
7.6 Hypotensive transfusion reaction
145
7.7 Acute haemolytic transfusion reaction
145
7.8 Delayed haemolytic transfusion reaction
146
7.9 Bacterial sepsis
146
7.10 Post-transfusion purpura
147
7.11 Transfusion-associated circulatory overload
147
7.12 Transfusion-related acute lung injury
148
7.13 Transfusion-associated dyspnoea
149
7.14 Transfusion-associated graft-versus-host disease
149
7.15 Iron overload / haemosiderosis
150
Page 4
Transfusion Medicine Handbook 3rd Edition
7.16 Transfusion-related immunosuppression
150
7.17 Transfusion-transmitted infection
150
7.18 Other infectious agents
151
7.19 Adverse event data
151
7.20 Other complications
152
8. CLINICAL ALTERNATIVES AND APPLICATIONS
153
8.1 Autologous blood collection and transfusion
153
8.2 Non-blood plasma volume expanders
154
8.3 Oxygen carrying compounds
155
8.4 Haemopoietic growth factors
155
8.5 Recombinant coagulation factors
157
8.6 Desmopressin acetate (Octostim®, Minirin®) 157
8.7 Tranexamic acid (Cyklokapron®) 159
8.8 Iron supplementation
160
9. OTHER SERVICES PROVIDED BY NZBS
162
9.1 Therapeutic apheresis
162
9.2 Therapeutic venesection
162
9.3 Tissue Bank
163
9.4 Autologous serum eye drops
163
9.5 Reference Laboratory (Immunohaematology)
164
9.6 Tissue Typing Laboratory
164
10. NZBS SAMPLE REQUIREMENTS
166
Transfusion Medicine Handbook 3rd Edition
Page 5
ABBREVIATIONS AND GLOSSARY
ABG
Arterial blood gas
ACE
Angiotensin-converting enzyme
ACS
Acute coronary syndrome
ACT
Activated clotting time
ADAMTS-13
A disintegrin and metalloproteinase with a thrombospondin
type 1 motif, member 13
ADP
Adenosine diphosphate
Adverse event
Untoward occurrence associated with the collection, testing,
processing, storage and distribution of blood components
and fractionated products. Serious adverse events are those
which may lead to death or to life-threatening, disabling or
incapacitating conditions for donors/patients, or which result
in, or prolong, hospitalisation or morbidity.
Adverse reaction
Untoward response in a donor or patient associated with the
collection or transfusion of blood components and fractionated
products. Serious adverse reactions are those which may
lead to death or to life-threatening, disabling or incapacitating
conditions or which result in, or prolong, hospitalisation or
morbidity.
AHF
Antihemophilic factor
AHTR
Acute haemolytic transfusion reaction
AIDS
Acquired immunodeficiency syndrome
AIHA
Autoimmune haemolytic anaemia
ALI
Acute lung injury
Allogeneic blood
Collected from one individual and intended for use by another
individual (as in allogeneic blood donation).
ANH
Acute normovolaemic haemodilution
Anti-HBs
Antibody to hepatitis B surface antigen (HBsAg). The presence
of anti-HBs is generally interpreted as indicating recovery and
immunity from hepatitis B virus (HBV) infection. Anti-HBs also
develops in a person who has been successfully vaccinated
against hepatitis B.
ANZSBT
Australian and New Zealand Society of Blood Transfusion
APTT
Activated partial thromboplastin time
ARCBS
Australian Red Cross Blood Service
ARDS
Adult respiratory distress syndrome
ASH
American Society of Hematology
ASTH
Australasian Society of Thrombosis and Haemostasis
ATG
Anti-thymocyte globulin
Autologous blood Collected from an individual and intended for their own use (as
in autologous blood donation).
Page 6
Transfusion Medicine Handbook 3rd Edition
ATIII
Antithrombin III
BCSH
The British Committee for Standards in Haematology
Blood component A therapeutic constituent separated from human blood (red
cells, platelets, fresh frozen plasma, cryoprecipitate and white
cells).
Blood (fractionated) A therapeutic protein fraction prepared from large pools
product
of human plasma under pharmaceutical conditions, e.g.,
coagulation factors, albumin, immunoglobulins.
BMI
Body mass index
BMS
Blood management system
BNP
B-type natriuretic peptide
BU
Bethesda unit
CHAD
Cold haemagglutinin disease
CMO
Chief Medical Officer
CMV
Cytomegalovirus is a member of the herpesvirus family,
transmissible by transfusion, and which may cause disease in
immunosuppressed patients.
CNS
Central nervous system
CoE
Council of Europe
CPAP
Continuous positive airways pressure
CPD / CPDA
Citrate phosphate dextrose and citrate phosphate dextrose
adenine anticoagulant solutions used when collecting blood
donations.
CSL
Compound sodium lactate
CSL Behring
A manufacturer of plasma therapies whose name derives
from the merger of several parent companies including CSL
(Commonwealth Serum Laboratories) Limited and ZLB
Behring.
CVP
Central venous pressure
DAT
Direct antigobulin (Coombs) test
DDAVP
1-desamino-8-d-arginine vasopressin (Desmopressin)
DHB
District Health Board
DHTR
Delayed haemolytic transfusion reaction
DIC
Disseminated intravascular coagulation
DNA
Deoxyribonucleic acid
DSTR
Delayed serologic transfusion reaction
ECG Electrocardiogram
ECMO
Extracorporeal membrane oxygenation
EDTA
Ethylenediaminetetraacetic acid
eGFR
Estimated glomerular filtration rate
EPO Erythropoietin
Transfusion Medicine Handbook 3rd Edition
Page 7
FBC
Full blood count
FEIBA
Factor VIII inhibitor bypassing activity
FFP
Fresh frozen plasma
FIX
Factor IX
FMH
Fetomaternal haemorrhage
FNAIT
Fetal and neonatal alloimmune thrombocytopenia
FNHTR
Febrile non-haemolytic transfusion reaction
Fractionated
A therapeutic protein fraction prepared from large pools
product
of human plasma under pharmaceutical conditions, e.g.,
coagulation factors, albumin, immunoglobulins.
FVIII
Factor VIII
FXIII
Factor XIII
G-CSF
Granulocyte-colony stimulating factor
GP Glycoprotein
GVHD
Graft-versus-host disease
HAE
Hereditary angioedema
HBsAg
Hepatitis B surface antigen; a serologic marker on the surface
of hepatitis B virus (HBV). It can be detected in high levels
in serum during acute or chronic hepatitis. The presence of
HBsAg indicates that a person is infectious.
HBV
Hepatitis B virus
HCV
Hepatitis C virus
HDFN
Haemolytic disease of the fetus and newborn
HES
Hydroxyethyl starch
HFE
High Iron Fe gene; mutations of HFE are responsible for
genetic haemachromatosis.
HLA
Human leucocyte antigen
HPA
Human platelet antigen
HPC
Haematopoietic progenitor cell
HIT
Heparin-induced thrombocytopenia
HIV
Human immunodeficiency virus
HSCT
Haematopoietic stem cell transplant
HTC
Hospital transfusion committee
HTLV
Human T-cell lymphotrophic virus
HUS
Haemolytic uraemic syndrome
ICH
Intracranial haemorrhage
IgA
Immunoglobulin A
IgG
Immunoglobulin G
IgM
Immunoglobulin M
INR
International normalised ratio
Page 8
Transfusion Medicine Handbook 3rd Edition
ITP
Immune thrombocytopenic purpura
IVIg
Intravenous immunoglobulin
IU
International units
IUT
Intrauterine transfusion
JVP
Jugular venous pressure
LDH
Lactate dehydrogenase
MBOS
Maximum blood order schedule
MTP
Massive transfusion protocol
MO
Medical Officer
NBA
Australian National Blood Authority
NHI
National Health Index
NHMRC
National Health and Medical Research Council of Australia
NOAC
Non-vitamin K-dependent oral anticoagulant
NSAID
Non-steroidal anti-inflammatory drug
NZBS
New Zealand Blood Service
NZRC
New Zealand Resuscitation Council
PAS
Platelet additive solution; nutrient media used in place of
plasma for platelet storage.
PBSC
Peripheral blood stem cell
PCC
Prothrombin complex concentrate
PCI
Percutaneous coronary intervention
PHARMAC
Pharmaceutical Management Agency of the New Zealand
Government
PID
Primary immunodeficiency diseases
PLT
Single therapeutic dose of platelets
PT
Prothrombin time
PTP
Post-transfusion purpura
RAADP
Routine antenatal anti-D prophylaxis
RANZCOG
Royal Australian and New Zealand College of Obstetricians
and Gynaecologists
RCNA
Royal College of Nursing of Australia
RCo
Ristocetin cofactor
Resuspended
Red cells from which the majority of plasma has been removed
red cells
and replaced with a nutrient or preservative solution, e.g.,
saline, adenine, glucose, mannitol (SAG-M)
RhD
RhD red cell antigen
RNA
Ribonucleic acid
SCIg
Subcutaneous immunoglobulin
SED
Serum eye drops
Transfusion Medicine Handbook 3rd Edition
Page 9
SHOT
Serious Hazards of Transfusion; UK haemovigilance
programme.
TACO
Transfusion-associated circulatory overload
TAD
Transfusion-associated dyspnoea
TA-GVHD
Transfusion-associated graft-versus-host disease
TMS
Transfusion Medicine Specialist
TNS
Transfusion Nurse Specialist
TPE
Therapeutic plasma exchange
TPO Thrombopoietin
TPR
Temperature/Pulse/Respiratory rate
TRAE
Transfusion-related adverse event
TRALI
Transfusion-related acute lung injury
TRIM
Transfusion-related immunomodulation
TTI
Transfusion-transmitted infection
TTP
Thrombotic thrombocytopenic purpura
vCJD
Variant Creutzfeldt-Jakob disease
vWF
von Willebrand factor
vWD
von Willebrand disease
VZV
Varicella zoster virus
Whole blood
Blood collected from a donor prior to separation into
constituent red cells, platelets and plasma.
WBCT
Whole blood clotting time
WBIT
Wrong blood in tube
WHO
World Health Organisation
Page 10
Transfusion Medicine Handbook 3rd Edition
FOREWORD
This handbook is designed to assist hospital staff and other health professionals in
modern transfusion medicine practice, particularly those who are prescribing and
administering blood products. In addition to information about the blood products
and services offered by the NZBS, it provides a framework for the clinical indications
for their use, the procedures for administration, and the management of adverse
reactions in patients.
The NZBS Clinical Compendium, NZBS Manufacturing Standards, hospital clinical
policies and other departmental manuals cover aspects of these guidelines in more
detail. The contents of the NZBS Clinical Compendium are also available on the
NZBS website (www.nzblood.co.nz). If information in these should not accord with
the principles outlined in this document, the differences should be referred in writing
to the New Zealand Blood Service, attention National Medical Director:
The National Medical Director
New Zealand Blood Service
Private Bag 92071
Auckland 1142
Telephone (09) 523 5733
Facsimile (09) 523 5754
The assistance of Transfusion Medicine Specialists and Transfusion Nurse Specialists in
the review of these guidelines is gratefully acknowledged. Comments and suggestions
for improvements in future editions are invited and should be addressed to the National
Medical Director.
If assistance is required for the management of transfusion support for patients in
particular clinical circumstances further information and advice can always be obtained
from NZBS Transfusion Medicine Specialists.
Transfusion Medicine Handbook 3rd Edition
Page 11
1
INTRODUCTION
1.1
Audience
Many people play an essential part in ensuring that the right blood components and
products are given to the right patient at the right time. This handbook is therefore intended
for all staff responsible for prescribing, supplying and administering blood components
and fractionated products. They include:■
■
Registered medical practitioners, nurses and midwives who assess patients
and who prescribe and order blood components and fractionated products to
be transfused.
■
Phlebotomists and others who collect and send pretransfusion samples.
■
Laboratory staff who ensure that blood components are compatible for
transfusion.
■
Orderlies and other personnel who deliver blood components and fractionated
products to hospital wards and clinics where patients are transfused.
■
Nurses and other clinical staff who check that, before being administered, the
supplied blood components and fractionated products are intended for the
identified patient, and who then observe the patient during and after the transfusion.
■
Medical and nursing students involved in any of the above activities.
■
Telephone operators who may have to make vital contacts in an emergency.
1.2
Evidence
Correctly used, transfusion can save lives and provide numerous clinical benefits.
However, the effectiveness of many current transfusion practices has not been
rigorously proven by clinical trials. It is not possible to offer complete evidence-based
guidelines for practice. Where good evidence is not available, the contents of this
handbook reflect best efforts to give a balanced view of current opinion about the
clinical practice of transfusion for patients in New Zealand.
1.3
Clinical Practice Guidelines
Australasian guidelines are available for the administration and appropriate use of blood
components and fractionated products. These have been developed by the Australian
& New Zealand Society of Blood Transfusion (ANZSBT) and the Australian National
Blood Authority (NBA) and supported by specialist colleges and medical specialists
of both Australia and New Zealand.
The NBA Patient Blood Management Guidelines, approved by the Australian National
Health and Medical Research Council (NHMRC), cover the use of blood components
(red blood cells, platelets, fresh frozen plasma, cryoprecipitate) and are available from
the NBA website at www.blood.gov.au/pbm-guidelines.
A summary of transfusion guidelines can be found on the reverse of the NZBS
Request for Blood Components or Products form and are presented in more detail in
Chapter 4: Blood Components. Other sources of information that may prove useful
are listed in Table 1.1.
Page 12
Transfusion Medicine Handbook 3rd Edition
However, the most important factors driving the success of moves toward improved
clinical quality reside within New Zealand hospitals (both DHB and private). Evidence
from the New Zealand National Haemovigilance Programme and similar programmes
overseas, such as the UK’s Serious Hazards of Transfusion scheme (SHOT), shows
that most serious errors relating to transfusion practice arise from administrative and
clerical errors, most of which are avoidable.
Table 1.1 Sources of Information
Organisation
Website
New Zealand Blood Service
www.nzblood.co.nz
NZBS Blood Resource
www.clinicaldata.nzblood.co.nz/
resourcefolder
Australian Red Cross Blood Service
www.transfusion.com.au
Australian National Blood Authority
www.blood.gov.au
Australian and New Zealand Society of
www.anzsbt.org.au
Blood Transfusion (ANZSBT)
Joint UK Blood Transfusion and Tissue
www.transfusionguidelines.org.uk
Services Professional Advisory Committee
British Committee for Standards in
www.bcshguidelines.com
Haematology (BCSH)
UK Serious Hazards of Transfusion scheme
www.shotuk.org
(SHOT)
NZ Ministry of Health
www.moh.govt.nz
New Zealand legislation
www.legislation.govt.nz
1.4 Haemovigilance
Blood components and fractionated products are biological in nature and carry
inherent risks in respect of infection or reactions in the recipient. Common and, for
the most part, minor reactions include febrile non-haemolytic and allergic transfusion
reactions. A small number of problems account for the majority of difficulties and
dangers associated with transfusion, for example delay in obtaining blood components
needed urgently, transfusing blood components and fractionated products intended
for another patient, over-transfusion leading to circulatory overload and pulmonary
oedema, and transfusion-transmitted bacterial infections. Fortunately, the risk of
transfusion-transmitted viral infections such as HIV, hepatitis B and hepatitis C is
relatively low when compared with other risks.
Clinical staff have the responsibility for recognising and reporting transfusion-related
complications to the blood bank, NZBS Transfusion Medicine Specialist/Medical Officer
or a Transfusion Nurse Specialist.
It is the task of NZBS and manufacturers of fractionated products to ensure that blood
supply and transfusion practice is as safe as possible. Similarly, local hospital blood
bank staff and the hospital transfusion committee should ensure that adverse events
are effectively investigated and reported.
Transfusion Medicine Handbook 3rd Edition
Page 13
Some Common Causes of Problems
Most of the problems associated with transfusion that cause delays and may put
the patient at risk are caused by poor communication, failure to follow documented
procedures and inadequately trained staff. The most frequently occurring problems are:
■
Prescribing blood components and fractionated products that are not required by
the patient or are not the most suitable treatment for the patient’s clinical condition.
■
Incomplete or inaccurate completion of request forms or sample tube labels.
■
Improper collection of samples possibly leading to ‘wrong blood in tube’ (WBIT)
incidents.
■
Delays caused by a failure to communicate accurately when and where blood
components and fractionated products are needed.
■
Transfusion of blood components and fractionated products that are intended
for another patient.
■
Failure to recognise and appropriately manage an adverse reaction occurring
during transfusion.
Notification and Investigation of an Adverse Transfusion Reaction
NZBS has produced a form for notifying the blood bank of the occurrence of a
transfusion reaction. Copies of the Notification and Investigation of Adverse Transfusion
Reaction form are normally available in each ward or from the blood bank.
On the reverse side of this form are guidelines for the management of adverse
transfusion reactions to assist clinical staff in the immediate care of the patient.
Haemovigilance Activities
NZBS is obliged to monitor the occurrence of adverse events during the transfusion
process from vein-to-vein (i.e., from donation collection through to transfusion), in line
with principles contained in the Council of Europe Guide to the Preparation, Use and
Quality Assurance of Blood Components. Donor incidents, specimen labelling errors,
blood bank errors, bedside checking errors and transfusion reactions are all examples
of activities which fall under the umbrella of ‘haemovigilance’ and for which NZBS has
systems in place to capture information.
In turn, the National Haemovigilance Programme specifically receives notification
of adverse events that occur during, or as a result of, transfusion. All New Zealand
hospitals and blood banks participate in the programme, reporting any transfusion-
related adverse events experienced by patients in the hospitals they serve. NZBS also
works with hospitals to ensure that adverse events are appropriately managed and
reported in a timely manner.
Transfusion is a complex process involving many different staff groups in the hospital.
Any failure in the transfusion chain has the potential to cause significant harm to, or even
death of, patients. Therefore all personnel involved in the transfusion process should
be encouraged to be vigilant and report any untoward events that they may observe.
Further information about the nature of adverse effects of transfusion and how they
should be reported to NZBS can be found in Chapter 7:
Adverse Events of Transfusion.
Page 14
Transfusion Medicine Handbook 3rd Edition
2
COLLECTION, TESTING AND PROCESSING OF BLOOD
DONATIONS
2.1
Blood Donors
Blood donors are essential to NZBS and the national blood supply. Up to 800 donors
are needed each working day to meet the country’s transfusion needs and the supply
of plasma for the manufacture of fractionated products. A great debt is therefore owed
to the many volunteer donors who so willingly help in this way.
There are five categories of blood donation:
■
Whole blood donation where a single unit of blood is collected. Whole blood
donors usually give about 470 mL of blood (with an additional 30 mL taken for
routine testing) and can donate up to four times per year.
■
Apheresis donation where plasma, platelets or white cells are specifically
collected from the donor’s whole blood using a machine known as a cell-
separator. Once these components are harvested, the donor’s red cells are
usually returned to them. Apheresis donors can donate more frequently than
whole blood donors and as often as every two weeks.
■
Pre-operative autologous donation where blood components are collected
from patients for transfusion to themselves during elective surgery. Refer to
Section 8.1:
Autologous Blood Col ection and Transfusion for further information.
■
Directed donation where one individual seeks to identify another individual
who will donate to provide blood components for either themselves or a close
family relative.
■
Selected donation where NZBS identifies donors to provide blood components
for recipients with rare blood types, or with red cell or platelet antibodies.
2.2
Donor Selection Criteria
The donor selection process contributes significantly to the safety of the donor pool
and the blood supply:
■
Whole blood donors are accepted from their 16th birthday up to their 66th
birthday. Established donors (those who have donated in the previous two years)
can donate up to their 71st birthday and thereafter up to their 76th birthday,
subject to annual medical review by an NZBS Medical Officer.
■
Donors must be in good health and weigh more than 50 kg. This ensures that the
process of donation is not detrimental to the donor’s health as well as protecting
the recipient from blood-borne infectious disease (such as bacterial sepsis, HIV,
hepatitis B and hepatitis C, malaria and variant Creutzfeldt-Jakob disease), donor
medications or other contaminants that could be harmful to them.
■
At each donation the donor completes a detailed health questionnaire and signs
a declaration that the health information provided is correct.
■
The donor’s haemoglobin level is checked each time they donate.
Transfusion Medicine Handbook 3rd Edition
Page 15
■
The donor’s completed medical history is evaluated by a suitably qualified health
professional and the donor accepted or deferred accordingly.
The NZBS website (www.nzblood.co.nz) provides detailed information on the current
donor eligibility and deferral criteria.
2.3
Self-Sufficiency and the Volunteer Status of Donors
New Zealand has adopted recommendations of the World Health Organisation (WHO)
in achieving self-sufficiency in safe blood supply, based on voluntary non-remunerated
blood donation (VNRBD).
Defined by the WHO, self-sufficiency means that the national needs of patients are met
in a timely manner, that patients have equitable access to transfusion services, blood
components and fractionated products, and that these are obtained from VNRBD of
national, and where needed, of regional origin, such as from neighbouring countries.
Donation is considered voluntary and non-remunerated if the person gives blood,
plasma or cellular components of his/her own free will and receives no payment for
it, either in the form of cash, or in kind which could be considered a substitute for
money. This would include time off work other than that reasonably needed for the
donation and travel. Small tokens, refreshments and reimbursements of direct travel
costs are compatible with voluntary, non-remunerated donation.
2.4
Informed Consent for Donation
Informed consent is a requirement of the
Code of Health and Disability Services
Consumer’s Rights (1996). Donors are required to be fully informed about the donation
process, the risks involved and their obligations as donors. The donor’s consent is
obtained when they complete the health questionnaire.
In consenting to donate, donors agree to the testing of their blood for blood group
and evidence of infectious diseases, as well as the use of their blood for transfusion,
teaching, diagnostic purposes or quality testing.
Information leaflets about the blood donation process and blood safety along with
a copy of the health questionnaire are available from NZBS blood centres and also
from the NZBS website.
2.5
Apheresis Donation
Apheresis is a procedure used to collect:
■
Plasma (plasmapheresis)
■
Cellular components (cytapheresis) including:
-
Platelets (plateletpheresis)
-
Granulocytes (granulocytapheresis)
-
Haematopoietic progenitor cells (HPCs) derived from peripheral blood
Red cells can also be collected by erythrocytapheresis (although NZBS does not
offer this service).
Page 16
Transfusion Medicine Handbook 3rd Edition
2.6
Directed and Selected Donation
When a person seeks to identify another individual who will donate blood to provide
blood components for either themselves or a close family relative, the practice is
termed a
directed donation. The request usually occurs within family relationships, in
particular parents of children, where there may be significant emotional anxiety reflecting
a concern regarding the perceived safety of a transfusion. There is no evidence that
directed donations either lead to improved patient care or reduce the risk of acquiring
transfusion-transmitted infections. As a result, NZBS does not support the practice of
directed donations and will discourage such requests. However, if a directed donation
is collected, the procedures for collecting, testing, storing, handling and transfusing
the unit must follow the procedures recommended for non-directed allogeneic blood
donations. For the purposes of directed blood donations only, the meaning of the
terms donor and donation refer to a volunteer who provides blood that is to be used
for a purpose specified by the person providing the blood, or to the derived blood
components, respectively. If a directed donation is not used for the purpose specified,
i.e., transfusion to the intended recipient, the donation and associated components
will be discarded and not made available for other recipients.
In some circumstances it may be necessary for NZBS to seek a compatible donor
from relatives. This practice is at the discretion of a Transfusion Medicine Specialist
and involves a
selected donation, not a directed donation. Donors are selected for
matched platelets, peripheral blood stem cells and rare blood groups.
Apheresis donors should normally meet the requirements for whole blood donation.
Exceptions to this must be authorised by a NZBS Transfusion Medicine Specialist/
Medical Officer. Such exceptions will normally only be made when the plasma or
platelets are of unusual therapeutic value and only when the NZBS Transfusion
Medicine Specialist/Medical Officer, who is aware of the health status of the donor,
has documented that the donor’s health permits apheresis donation.
2.7 Haemochromatosis
Genetic or hereditary haemochromatosis is mainly associated with a defect in the
HFE (High Iron Fe) gene.
HFE helps regulate the amount of iron absorbed from food.
There are two important mutations in
HFE, namely C282Y and H63D, of which C282Y is
the most important. When inherited from both parents, C282Y causes a variable increase
in iron absorption which, if untreated, may lead to iron overload and organ dysfunction.
Management consists of lifelong monitoring, avoidance of an iron-rich diet and,
depending on clinical phenotype, therapeutic venesection to deplete iron stores
followed by maintenance venesection to prevent recurrence of iron overload.
Patients who are clinically well and meet all blood donor acceptance criteria may be
enrolled as normal blood donors. NZBS checks the ferritin level periodically while iron
depletion is in progress. Once stable iron-deplete levels are achieved, annual monitoring
by the patient’s general practitioner is recommended.
2.8
Cord Blood Donation
NZBS currently does not routinely provide this service and all requests for the
col ection of directed cord stem cel s should be referred to a NZBS Transfusion
Medicine Specialist.
Transfusion Medicine Handbook 3rd Edition
Page 17
2.9
Testing of Donor Blood
Every unit of blood collected is tested for the following:
■
ABO and RhD blood groups
■
Red cell antibodies
■
Infectious diseases:
-
HIV (HIV Ag/Ab and HIV RNA)
-
Hepatitis B (HBsAg and HBV DNA)
-
Hepatitis C (anti-HCV and HCV RNA)
-
Syphilis (
Treponema pallidum antibodies)
Other testing restricted to specific donor groups and, where indicated, individual
donors or donations:
■
HTLV-I /II antibody on all new donors
■
CMV antibody on donations to be used as components for patients at high risk
from CMV infection
■
Plasmodium antibody on donations from malaria-risk donors
■
Trypanosoma cruzi antibody on donations from Chagas-risk donors
■
Ferritin on donors failing the haemoglobin screening test
■
Direct antiglobulin test (DAT)
■
Extended red cell antigen typing
■
HLA antibody on previously pregnant (> 20 weeks gestation) female plateletpheresis
donors
■
HLA (human leucocyte antigen) or HPA (human platelet antigen) genotyping
2.10 Leucodepletion
All blood components for direct clinical transfusion are leucodepleted. Leucodepletion
is a process for removing white cells (leucocytes) from blood components. This is
achieved by means of a special filter or by differential centrifugation. Leucodepleted
blood components should contain < 5 x 106 white cells per unit.
NZBS introduced universal prestorage leucodepletion in July 2001, initially as one of
a series of precautionary measures against the potential risk of transmission of variant
Creutzfeldt-Jakob disease (vCJD) by blood transfusion.
There is good evidence to support the value of leucodepletion in preventing transfusion-
associated transmission of some infectious agents and in reducing some of the adverse
immunomodulatory effects of allogeneic transfusion:
■
Reduction in frequency of febrile non-haemolytic transfusion reactions.
■
Prevention or delay of alloimmunisation to human leucocyte antigens.
Page 18
Transfusion Medicine Handbook 3rd Edition
■
Prevention or delay of platelet refractoriness due to alloimmunisation.
■
Reduction in the risk of CMV transmission.
Leucodepletion also has other theoretical advantages:
■
Reducing the risk of other leucocyte-associated blood borne infections, such
as transmission of HTLV-I/II, and inadvertent bacterial contamination of blood
components.
■
May reduce the risk of perioperative infection or cancer recurrence by reducing
the immunomodulatory effects of blood transfusion.
■
May prevent some cases of transfusion related acute lung injury (TRALI).
2.11 Processing of Collected Blood to Components
Blood donations are processed into various components including red cells, platelet
concentrates, fresh frozen plasma and cryoprecipitate. Blood components are prepared
at NZBS processing centres under strictly controlled manufacturing conditions.
Blood Component Labelling Blood components supplied for transfusion have a blood component label applied by
the manufacturer specific to each individual component type. These labels provide
important information for those who administer blood components and also allow the
origins of the component to be traced.
Labels should state the details of the component and its composition, the conditions
under which it can safely be stored and the date and time of expiry. Components from
single donors must carry the unique donation number that identifies the donation.
Compatibility Labels
A ‘compatibility label’ will be applied by the blood bank issuing the component to the
patient. This label uniquely identifies the patient for whom the component has been
selected. An essential bedside check before transfusing any blood component is to
make sure that the details on the compatibility label match exactly those of the patient.
2.12 Processing of Collected Blood to Fractionated Products
Fractionated products, also known as plasma derivatives, are partial y purified
therapeutic preparations of plasma proteins. They are manufactured in large-scale
pharmaceutical processes from large volumes of source plasma. For example, each
batch of Intragam®P typically uses 7.5 - 10 tonnes of plasma (from approximately
25,000 - 30,000 individual whole blood donations).
The plasma is processed by a variety of techniques classically involving Cohn
fractionation but more recently chromatographic techniques such as gel filtration, ion
exchange and affinity chromatography have been used.
Finally, the purified plasma undergoes specific virus removal or inactivation steps, for
example heat-treatment, solvent/detergent treatment, incubation at low pH or filtration.
The final products are supplied as freeze-dried powders or solutions.
All of the plasma obtained by NZBS through the collection of whole blood, together
with approximately 80% of the apheresis plasma, is forwarded to CSL Behring in
Melbourne for the manufacture of fractionated products.
Transfusion Medicine Handbook 3rd Edition
Page 19
Product labelling
Fractionated products are identified by both the carton packaging and labels of the
product container. Included with every product is an insert provided by the manufacturer
with detailed information concerning the product relating to its composition, indication
for use and administration.
Blood bank labelling
All fractionated products issued to a patient have a computer-generated label attached
that provides details of the patient and product batch number. This label is used at
the bedside check of the patient’s identity and then placed in the clinical notes as a
record of transfusion.
2.13 Blood Components and Fractionated Products as Medicines
Blood components and fractionated products are classified as prescription medicines
under New Zealand legislation. The
Medicines Act 1981, Medicines Regulations 1984,
Medicines Amendment Regulations 2011 and
Medicines Amendment Act 2013 provide
the legal framework under which blood components and fractionated products may
be manufactured and supplied as well as stipulating who may prescribe them. Under
the regulations, medical practitioners, registered midwives and nurse practitioners all
have prescribing rights for blood components and fractionated products in accordance
with their regulated scopes of practice.
Page 20
Transfusion Medicine Handbook 3rd Edition
3
GUIDE TO GOOD TRANSFUSION PRACTICE
Both blood components and fractionated products are biologic material and, in
the case of components containing blood cells, are living human tissues. They are
prescription medicines intended for use by medical practitioners and midwives in the
treatment of patients.
Blood transfusion therapy has had a central role in the advances and practice of modern
medicine. As in other areas of clinical medicine, prescribers need to consider both
the benefits and risks of blood transfusion. Professional judgement based on clinical
evaluation determines selection of blood components and fractionated products,
dosage, rate of administration and sometimes other decisions in situations not covered
in this general introduction to blood transfusion practice.
The presence of contaminants such as immunogenic cellular and protein elements,
viable donor cells and infectious agents in blood cannot be totally avoided and indeed
may cause undesirable side ef ects in some recipients. The information in this handbook
cannot therefore be considered or interpreted as an expressed or implied warranty
of the safety or fitness of the described blood components or fractionated products
when used for their intended purpose.
3.1
Clinical Governance
A quality management system is needed wherever transfusion therapy is practised.
In this context ‘quality’ includes adequate documentation of both the transfusion
process and its outcomes.
All institutions that transfuse blood components and fractionated products should
develop and maintain local policies and procedures that reflect best national and
international transfusion practice. Local policies and procedures should include
guidance on:
■
Informed consent.
■
Requesting blood components and fractionated products.
■
Collection of blood samples for pretransfusion compatibility testing.
■
Collection of blood components and fractionated products from the hospital
blood bank or other sites.
■
Delivery of blood components and fractionated products to where the transfusion
is to be given.
■
Administration of blood components and fractionated products.
■
Care and monitoring of patients receiving a transfusion.
■
Documentation of transfusion.
■
Management and reporting of adverse events.
■
Staff responsibilities and the training required for these procedures.
Transfusion Medicine Handbook 3rd Edition
Page 21
3.2
Prescribing Blood Components and Fractionated Products
Prescribing blood components and fractionated products is normally the responsibility
of a registered medical practitioner. Registered midwives and nurse practitioners
have limited prescribing rights as defined by the
Medicines Regulations 1984 and
Medicines Amendment Regulations 2011. For example, registered midwives are able
to prescribe prescription medicines but only in the course of antenatal, intrapartum
and postnatal care.
Decisions to transfuse should, taking individual patient needs into account, be based
on international and regional guidelines such as the Australian National Blood Authority
Patient Blood Management Guidelines.
It is the responsibility of the prescribing practitioner to ensure that transfusion therapy
is given only when clearly indicated and that the patient is appropriately monitored
during the transfusion procedure.
The following questions should be taken into consideration when deciding to transfuse:
■
What improvement in the patient’s condition am I aiming to achieve?
■
Can I minimise blood loss to reduce the patient’s need for transfusion?
■
Are there any other treatments I should give before making the decision to
transfuse?
■
What are the specific indications for transfusing this patient?
■
Do the benefits of transfusion outweigh the risks to this particular patient?
■
Has the patient been given a clear explanation of the potential risks and benefits
of blood transfusion therapy in his or her particular case?
Once the decision to transfuse has been made it is also important to consider the following:
■
Have I recorded my decision to transfuse and the reasons for transfusion on
the patient’s chart and completed any documentation used in the ordering or
administration of blood components or fractionated products?
■
Will a trained person monitor this patient and respond immediately if any acute
transfusion reactions occur?
■
Has crossmatching and other relevant testing been carried out?
Prescribing Unapproved Pharmaceuticals
Practitioners should, where possible, prescribe pharmaceuticals that are approved under
the Medicines Act 1981. If Practitioners are planning on prescribing an unapproved
pharmaceutical or a pharmaceutical for an indication for which it is not approved,
Practitioners should:
a) be aware of and comply with their obligations under Section 29 of the Medicines
Act 1981 and otherwise under that Act and the Medicines Regulations 1984;
b) be aware of and comply with their obligations under the Health and Disability
Commissioner’s Code of Consumer Rights, including the requirement to obtain
informed consent from the patient; and
Page 22
Transfusion Medicine Handbook 3rd Edition
c) exercise their own skill, judgement, expertise and discretion, and make their own
prescribing decisions with respect to the use of an unapproved pharmaceutical
or a pharmaceutical for an indication for which it is not approved.
Where medicines, supplied under Section 29 of the Medicines Act, are used for
emergency situations, the required patient details may be retrospectively provided to
the supplier.
3.3
Informed Consent to Receive a Blood Transfusion
Informed consent for transfusion is a requirement of the
Code of Health and Disability
Services Consumers’ Rights (a regulation under the
Health and Disability Commissioner
Act 1994). This requires that patients be provided with information and an explanation
of the purpose for which blood components and fractionated products are being
prescribed and that they consent to transfusion.
Patients or their relatives may be worried about the risks of transfusion. Although most
patients will be prepared to give consent for a transfusion after receiving appropriate
information, some will seek and require quite detailed reasons for the transfusion,
information on the risks involved and the alternatives available (such as autologous
transfusion).
Some patients may refuse transfusion for personal or religious reasons, for example
members of the Jehovah’s Witnesses faith. Some of these patients may be prepared
to accept fractionated products or other alternatives.
The seeking of informed consent, together with the reasons for the transfusion, should
be recorded in the clinical record of the patient.
NZBS provides a range of leaflets to support the process of gaining informed consent.
Copies of these should be available at all sites where blood may be transfused and
can also be obtained from NZBS Transfusion Nurse Specialists or the hospital blood
bank. This information is also available on the NZBS website (www.nzblood.co.nz).
3.4
Requesting Blood Components and Fractionated Products
The NZBS
Request for Blood Components and Products form is used in most New
Zealand hospitals and follows a prescription written in the patient clinical record. Blood
components and fractionated products are normally obtained from the hospital blood
bank and local hospital policies will determine which staff can place orders. It should
be noted that the form used to request blood components and fractionated products
is not a prescription.
The request form must:
■
Correctly identify the patient.
■
Provide details of any previous transfusion or obstetric history.
■
Indicate the quantity of the blood component or fractionated product required,
when it is required and where it should be sent to.
Full and accurate completion of the request form is essential for:
■
Ensuring that the right quantity and type of blood component or fractionated product
is made available to the right patient, at the right time, and in the right place.
Transfusion Medicine Handbook 3rd Edition
Page 23
■
Minimising the risk of patient identification errors.
■
Alerting the blood bank to the possibility of antibodies, based on a history of
previous transfusions or pregnancy, in which case suitably matched blood may
be required.
Patients who, at the time of admission, cannot be reliably identified must be given
an identity band with a unique number. This number must be used to identify this
patient until full and correct details are available and are properly communicated to
the blood bank.
3.5
Blood Stock Management: The Maximum Blood Order Schedule
The maximum blood order schedule (MBOS) is one way a blood bank manages blood
stock. A typical MBOS lists surgical and other interventional procedures along with the
number of red cell units normally required in preparedness for transfusion in association
with these procedures. The number of units is determined by analysis of historical
blood usage figures. Many laboratories formulate a MBOS based on local surgical
experience, while others adopt a generic schedule. An example of such a schedule
is available in the 2016 Australian and New Zealand Society of Blood Transfusion
(ANZSBT)
Guidelines for Transfusion, Pre and Postnatal Immunohaematology Testing
(www.anzsbt.org.au).
The goal of the MBOS is to promote efficient use of red cells. It provides guidance to
(junior) medical staff on the number of units of red cells likely to be required for various
surgical procedures and is also a valuable guide for the blood bank, particularly those
which routinely perform serological crossmatching (generally smaller facilities). For
procedures where red cells are seldom required, the MBOS will simply recommend
that no units should be crossmatched and a group and screen performed.
If more red cells are ordered for a patient than required or they are held crossmatched
unnecessarily, then these units may be unavailable for other patients and there is a
chance that the red cells will expire before being used. The clinician therefore needs
to have a valid rationale to order more units than is mandated by the MBOS.
If the patient has a positive antibody screen or special transfusion requirements the
blood bank may adjust the number of red cells crossmatched (from what is specified
in the MBOS) to avoid potential problems should transfusion be required.
Together, the MBOS, regular monitoring of crossmatch-to-transfusion (C:T) ratios
for individual surgical procedures and adoption of a ‘group and screen’ policy are
all helpful in ensuring maximum use of the available stock of red cell components.
3.6
Collecting Blood Samples for Pretransfusion Testing
Correct identification of the patient before collecting the pretransfusion sample is vital
in avoiding ‘wrong blood in tube’ (WBIT) episodes. Only when the patient’s identity
is positively confirmed can the request form be completed, blood sample taken and
sample tube labelled.
Sample collection must be performed in accordance with hospital policy (or the
laboratory manual) and observe the following principles:
■
At the time of taking the sample ask the patient (if conscious or rational) to state
their given name(s), family name and date of birth.
Page 24
Transfusion Medicine Handbook 3rd Edition
■
Check this information against the patient’s identification bracelet and details
on the request form to make sure that the details are identical.
■
Collect the blood sample into the correct sample tube.
■
Hand label the sample* with the following information:
-
Patient’s family name
-
Patient’s given name(s)
-
Patient’s NHI number
-
Patient’s date of birth
-
Signature or initials of the collector
-
Date and time sample collected
* Please note: The information on the request form will be checked against that on the sample tube
and both must be identical. NZBS Blood Bank does not accept pretransfusion samples labelled
with pre-printed labels.
■
Sample tubes must be hand labelled in the presence of the patient, at the time
the blood is collected, by the person who obtained the sample:
-
Sample labelling must never be delegated to a third party.
-
Sample tubes must not be prelabelled before the sample is obtained
because of the risk of bleeding the patient’s blood into the wrong tubes.
-
Sample tubes must never be labelled away from the patient’s presence
because of the risk of labelling the sample tube and/or request form with
another patient’s details.
■
In the case of an unconscious (or irrational) patient, alternative identification
procedures may be necessary, for example verification by the next of kin, as
well as checking the identification wristband.
■
The sample acceptance criteria used by NZBS are based on ANZSBT
Guidelines
for Transfusion, Pre and Postnatal Immunohaematology Testing. Blood Bank staff
are not authorised to accept samples which do not meet labelling requirements.
Where necessary these will be rejected and new samples requested. Where a
dispute arises in relation to a sample, the final decision on suitability for testing
will lie with an NZBS Transfusion Medicine Specialist/Medical Officer.
3.7
Pretransfusion Testing
Using the pretransfusion blood sample taken from the patient, the blood bank will
perform a ‘group and screen’ (sometimes also referred to as ‘group and hold’,
‘group and save’ or ‘type and screen’). A ‘group and screen’ consists of the following
procedures:
■
ABO and RhD group.
■
Antibody screen.
■
Checking for previous or duplicate records for the patient and, when these are
available, comparing current results with historical findings.
Transfusion Medicine Handbook 3rd Edition
Page 25
If red cells are subsequently required the blood bank will select appropriate donor
units and perform a final compatibility check.
Various approaches exist for compatibility testing. This may involve either a serological
crossmatch of the patient’s plasma versus donor red cells or, in some hospitals, an
electronic crossmatch, where the blood bank computer performs the last compatibility
check. Once pretransfusion testing is completed blood can be issued to the ward
or operating theatre.
3.8
Patients with a Positive Antibody Screen
If the patient is found to have a clinically significant red cell antibody during antibody
screening, blood that does not have the corresponding antigen will be needed for
transfusion. The relevant antigen-negative donor blood is selected and crossmatched
against the patient’s plasma. This can be a time-consuming process but is necessary
if the patient is to receive compatible blood.
If there is insufficient time for full identification of the antibody or to obtain antigen-
negative units, transfusion of (potentially) incompatible blood may be recommended.
In these circumstances a NZBS Transfusion Medicine Specialist/Medical Officer will
contact the clinical staff to discuss the comparative risks of delaying transfusion versus
transfusing potentially incompatible blood.
3.9
Sample Validity (‘72-Hour Rule’)
The sample validity period, during which time a pretransfusion sample may be held
and used to provide crossmatched blood, depends on the patient’s transfusion or
obstetric history.
Red cell antibodies can rapidly appear in response to stimulation by transfused red
cells or as a result of pregnancy. Consequently, if in the three months preceding
collection of the pretransfusion sample the patient has been transfused with red cells
(or platelets), is currently pregnant (or has been), the sample will have a validity of 72
hours. This is known as the ‘72-hour rule’.
Longer expiry times are applied if the patient is known not to have a history of
transfusion or pregnancy.
Table 3.1: Sample Validity Criteria
Patient History
Sample Expiry
Patient transfused or pregnant in previous 3 months (or
72 hours
history not known) or has history of clinically significant red cell
antibodies
Patient neither transfused nor pregnant in last 3 months and no
7 days
history of clinically significant red cell antibodies
Preadmission sample and patient neither transfused nor
21 days
pregnant in last 3 months and no history of clinically significant
red cell antibodies
For preadmission samples, the requirement for 21-day expiry must be clearly noted
on the request form along with the proposed date of surgery or procedure. Details of
Page 26
Transfusion Medicine Handbook 3rd Edition
whether or not the patient has a transfusion and/or obstetric history must also be given.
Failure to provide this information will result in the sample being given a 72-hour expiry.
Once a transfusion episode has commenced the pretransfusion sample, and any
associated results, will cease to be valid either at the original expiry of the sample, or
72 hours from when transfusion of the first unit commenced, whichever eventuates first.
If further transfusions are necessary a new sample will be required. Each new sample
will have an expiry of 72 hours until a gap of three months between transfusions (or
since pregnancy) has occurred.
Extensions to sample expiry times may be possible at the discretion of a NZBS
Transfusion Medicine Specialist/Medical Officer. If you have any queries regarding
sample validity contact the local blood bank for advice.
3.10 Provision of Red Cells in an Emergency
The nature and availability of red cells in an emergency situation depends on the
urgency with which blood is required and the extent of pretransfusion testing that can
be completed within the appropriate response time.
Table 3.2: Availability of Red Cell Units
Tests Completed
Units Selected
Availability
None
Emergency O RhD negative blood
Immediate
(uncrossmatched)
Limited testing
ABO/RhD group specific blood
15 minutes
(ABO/RhD type only)
(uncrossmatched)
All testing
ABO/RhD group specific and
45 minutes1
(Full ‘Group and Screen’)
compatible blood
1Assuming a negative antibody screen.
If red cells are required immediately and before pretransfusion testing can be
completed, the use of emergency group O RhD negative units should be considered.
As stocks of group O RhD negative red cells are limited, a timely switch to red cells of
the patient’s group is recommended. It should be noted that whilst emergency red cells
are group O RhD negative, complete serological compatibility cannot be guaranteed
as the patient may have a red cell antibody.
Where time allows a confirmed ABO/RhD type to be obtained, uncrossmatched blood
of the same group as the patient will be provided. On completion of a full group and
screen, and as long as the patient does not have a red cell antibody, group-specific
and crossmatch-compatible blood is available.
If the patient has a red cell antibody, finding compatible blood may be delayed while
the antibody is identified and antigen-negative red cells selected and crossmatched.
Under these circumstances, if blood is still urgently required, incompatible red cells
may have to be used. However the decision to transfuse should be made in discussion
with a NZBS Transfusion Medicine Specialist/Medical Officer.
Transfusion Medicine Handbook 3rd Edition
Page 27
3.11 Removal from Storage and Time Limits for Infusion
It is important that blood components are transfused as soon as possible following
receipt from the blood bank so that the required efficacy is achieved and unwanted
bacterial proliferation is avoided.
Red Cells
■
Transfusion should begin as soon as possible following removal of the unit from
a monitored blood refrigerator or validated transport container.
■
Transfusion of red cells should be completed within four hours of removal from
a monitored blood refrigerator or validated transport container.
■
Where a short delay occurs (or is anticipated) before starting a transfusion,
red cells may be held at ambient temperature provided the transfusion can be
completed within four hours of the blood being issued from a monitored blood
refrigerator.
■
If transfusion cannot be started within 30 minutes, the unit should normally be
returned without delay to a monitored blood refrigerator for controlled storage.
■
Red cells must be stored in a refrigerator that is manufactured and validated for
the purpose of storing red cell components and has permanent temperature
monitoring. They must not be stored in a ward refrigerator, domestic refrigerator
or refrigerator intended for vaccine storage.
■
If a unit of red cells has been out of controlled storage for more than 30 minutes
and there is no prospect of imminent transfusion it should be returned to the blood
bank for disposal. The unit cannot be accepted back into blood bank stock.
Platelets
■
Transfusion should begin as soon as the platelets are received from the blood
bank.
■
Transfusion of platelets should be completed within one hour of issue from the
blood bank.
■
If not used immediately, platelets must be returned to the blood bank and
controlled storage within one hour of issue.
■
Platelets are stored (usually in the blood bank) at room temperature 20 - 24ºC
with constant agitation.
■
Platelets must not be transported or stored in a refrigerator or chilled transport
container.
■
If platelets have been out of controlled storage for more than one hour,
acceptance back into blood bank stock is conditional on evidence of suitable
storage.
Fresh Frozen Plasma
■
Transfusion should begin as soon as the thawed plasma is received from the
blood bank.
Page 28
Transfusion Medicine Handbook 3rd Edition
■
Transfusion of plasma should be completed within four hours of thawing.
■
If the plasma is not going to be used or transfusion cannot be started within
30 minutes it must be returned to the blood bank immediately. If returned within
30 minutes of issue then it can be stored for up to 24 hours at 2 - 6°C during
which time it may be reissued to the same or a different patient. If not used
within 24 hours, the returned plasma expires.
■
If the plasma has been out of controlled storage for more than 30 minutes it
cannot be accepted back into blood bank stock.
■
Once thawed, plasma must not be refrozen.
Cryoprecipitate
■
Transfusion should be started as soon as the thawed cryoprecipitate is received
from the blood bank.
■
Transfusion of cryoprecipitate should be completed within four hours of thawing.
■
If the cryoprecipitate is not going to be used or transfusion cannot be started
within 30 minutes it must be returned to the blood bank immediately, where
it can then be stored for up to 4 hours at ambient (room) temperature during
which time it may be reissued to the same or a different patient. If not used
within 4 hours, the returned cryoprecipitate expires.
■
If the cryoprecipitate has been out of controlled storage for more than 30 minutes
it cannot be accepted back into blood bank stock.
■
Once thawed, cryoprecipitate must not be stored in the refrigerator nor should
it be refrozen.
3.12 Administration and Observation of Transfusion
All transfusions should be performed and monitored in accordance with relevant
hospital policies and guidelines. The 2011 ANZSBT & Royal College of Nursing of
Australasia (RCNA)
Guidelines for the Administration of Blood Products may provide
supplementary guidance.
■
Before transfusion always check the identity of the recipient, that the correct
blood component or fractionated product has been obtained, and that it has
not expired.
■
Transfuse only if the patient can be observed and monitored by trained staff.
3.13 Rate of Infusion and Precautions
The appropriate rate of transfusion may vary significantly according to the clinical
circumstances:
■
Patients who are actively bleeding and/or are in hypovolaemic shock will require
blood components to be transfused as rapidly as possible.
■
Patients with cardiac failure are at risk of circulatory overload and it will
be necessary to transfuse slowly and cautiously with frequent monitoring.
Concomitant use of diuretics should also be considered.
Transfusion Medicine Handbook 3rd Edition
Page 29
■
For fractionated products, the package insert provides guidance on specific
protocols regarding the administration of the product.
Advice must be sought from the doctor responsible for the patient if there is any
doubt about the way or how rapidly a blood component or fractionated product
should be transfused.
3.14 Infusion Pumps
Approved infusion pump devices may be used to assist transfusion. Check the
manufacturer’s instructions before using a pump to transfuse red cells or platelet
concentrates.
3.15 Blood Administration Sets and Filters
All blood components, including platelets and plasma components, must be transfused
through a standard sterile blood administration set incorporating a suitable integral
screen filter (normally 170 - 200 micron pore size). The filter is designed to trap cellular
aggregates, cellular debris and clots potentially harmful to the patient. Microaggregate
filters are not indicated.
In New Zealand, bedside leucocyte-depleting filters are not required as all blood
components undergo prestorage leucodepletion during processing.
Blood administration sets must be used in accordance with the manufacturer’s
instructions and hospital policy. The following provides a general guide to the use of
blood administration sets.
Administration sets may be primed with 0.9% sodium chloride (‘normal saline’) or
the component being transfused. Compound sodium lactate (Hartmann’s or Ringer-
Lactate), Plasmalyte in 5% glucose and dextrose solutions must not be used.
■
Because of the risk of bacterial proliferation, each administration set should only
be used for up to 12 hours of transfusion or until the filter becomes clogged.
■
One administration set may be used for transfusing up to 4 red cell units provided
the flow rate remains adequate.
■
In a massive transfusion setting, 8 - 10 units may be transfused before the set
is changed, provided the flow rate remains adequate without evidence of filter
clogging and the set is changed at least every 12 hours.
■
Transfuse platelets through a fresh administration set. Transfusing platelets
through an administration set previously used for red cells is not recommended.
■
Administration sets should be flushed with normal saline before and after platelet
transfusion if the same set is to be subsequently used for the transfusion of red
cells or FFP.
■
If there is doubt about the appropriateness of filters or their use, contact the
blood bank, NZBS Transfusion Medicine Specialist/Medical Officer or NZBS
Transfusion Nurse Specialist.
3.16 Warming of Blood Products
If warming is clinically indicated, use only an appropriate and approved system. The
warming system must be equipped with a visible thermometer and an audible alarm
Page 30
Transfusion Medicine Handbook 3rd Edition
as malfunction can result in red cell haemolysis. Blood components must not be
warmed above 41°C.
Clinical indications for the use of blood warmers:
■
Large volumes transfused rapidly, for example > 50 mL/kg/hr in adults and
> 15 mL/kg/hr in children.
■
Neonatal exchange transfusions.
■
Trauma situations in which core-rewarming measures are indicated.
■
Patient rewarming phase during cardiopulmonary bypass surgical procedures.
■
Transfusions for patients with clinically significant cold reactive antibodies (‘cold
agglutinins’), i.e., symptomatic cold haemagglutinin disease (CHAD).
Blood warmers are not indicated for routine transfusion of blood. Blood warming is
seldom necessary or desirable for elective transfusion at conventional rates, even for
patients with asymptomatic cold agglutinins.
3.17 Compatible Intravenous Solutions
■
No drugs or additives, other than 0.9 % sodium chloride (‘normal saline’)
intravenous infusion, are recommended to be mixed with red cells before or
during transfusion.
■
Do not use Plasmalyte 148 in 5% glucose or 5% dextrose solutions as these
may induce haemolysis.
■
Do not use Gelofusal® or compound sodium lactate solutions, e.g., Hartmann’s
or Ringer-Lactate, as these contain calcium and may induce clot formation in
the blood component bag and/or administration set.
3.18 Adding Medication to Blood Components
Do not add medication to any blood component unless specifical y approved by
hospital policy. If there is doubt, contact a NZBS Transfusion Medicine Specialist/
Medical Officer or NZBS Transfusion Nurse Specialist for advice.
3.19 Documentation of Transfusion
Full and accurate documentation of every step of the transfusion process is vital. Not
only is this important for effective investigation of serious transfusion-related adverse
events such as transfusion-transmitted infections, it also facilitates auditing of al
aspects of the transfusion process and is essential in ensuring vein-to-vein traceability of
blood components and fractionated products. Table 3.1 summarises the requirements
of documentation associated with transfusion.
The majority of transfusion errors are administrative or clerical. Common errors include:
■
Failure to identify the patient properly when taking pretransfusion blood samples.
■
Failure to correctly label the patient’s pretransfusion blood sample at the bedside.
■
Transcription errors in the laboratory.
■
Failure to clearly identify the intended recipient prior to transfusion.
Transfusion Medicine Handbook 3rd Edition
Page 31
Table 3.1: Documentation of Transfusion
Requirement
Action
Prescription
Constitutes the legal instruction (from the medical practitioner,
registered midwife or nurse practitioner) to administer blood
components and fractionated products and must be placed
in the patient’s medical record. The prescription must be
legibly written, be dated and include the prescriber’s name
and signature. The prescription must specify:
■
blood component / fractionated product to be administered
■ quantity
■
route of administration
■
rate of infusion
■
any special requirements
Justification for
Document the transfusion decision (based on recognised
transfusion
clinical practice guidelines) in the patient’s medical record.
Informed consent
Must be obtained from the patient and documented (in the
medical record) except in emergencies where appropriate
communication is not possible.
Patient’s medical
■
Prescription of the blood component / fractionated product.
record
■
Evidence of informed consent.
■
The compatibility label (or other labelling with patient and
blood component / fractionated product details), which
should be removed after completing the transfusion and
attached to the transfusion record.
■
Evidence of the bedside checking procedure.
■
Vital signs during the transfusion.
■
The indication for the use of the blood component /
fractionated product.
■
The date of transfusion and the time transfusion started
and finished.
■
The number and type of blood components / fractionated
products (including the donation / batch numbers).
■
Whether or not the desired effect was achieved.
■
A record of the occurrence and management of any
transfusion-related adverse event.
Retention of
■
In accordance with statutory requirements, namely the
records
Health (Retention of Health Information) Regulations 1996,
amended by section 111(2) of the
New Zealand Public
Health and Disability Act 2000, records of transfusion
must be retained for a minimum of 10 years.
3.20 Local Systems and Procedures
It is important to be familiar with local hospital policies in regard to transfusion practices.
Visiting the blood bank may also be useful in gaining an appreciation of the local system
for obtaining blood components and fractionated products and meeting the clinical,
nursing and scientific staff who provide transfusion services. There is no substitute for
talking with people who are working to help you care for your patients.
Page 32
Transfusion Medicine Handbook 3rd Edition
3.21 Reporting of Adverse Events
If the patient experiences a reaction as a result of transfusion, has received an
inappropriate transfusion, received a blood component or fractionated product
intended for another patient or where special requirements (for example, irradiated
components) are not met, these are regarded as transfusion-related adverse events.
Transfusion reactions can cause a patient’s condition to rapidly deteriorate with
respiratory distress, hypotension and collapse. Any signs or symptoms suggesting a
reaction should not be ignored, but rather assessed immediately.
Any untoward reaction or event occurring during or after the transfusion must be
reported immediately to the blood bank and serious events (life-threatening or with
a risk of major morbidity) should also be reported to a NZBS Transfusion Medicine
Specialist/Medical Officer or NZBS Transfusion Nurse Specialist.
Adverse effects of transfusion and the reporting of events is covered in detail in Chapter
7:
Adverse Effects of Transfusion.
Transfusion Medicine Handbook 3rd Edition
Page 33
4
BLOOD COMPONENTS
Blood components are made from blood donations and may involve one or more
simple physical processing steps to separate the constituents of blood.
The entries in this section are summaries of information presented in the NZBS
Clinical
Compendium, which is a source of reference for the clinical activities of NZBS. The
compendium provides a framework for clinicians on the nature, composition and clinical
use of the components and contains detailed individual component descriptions,
quality specifications, storage requirements, dosage and administration guidelines,
precautions and contraindications to use, along with information on potential adverse
reactions.
The compendium is a controlled document. Regular updates of the individual
documents it contains are produced and disseminated to holders. Documents within
the compendium can also be found in the
Clinical Information section of the NZBS
website (www.nzblood.co.nz).
4.1
ABO Blood Groups and Antibodies
ABO is the most important of the human blood group systems. There are four different
ABO blood groups, determined by whether or not an individual’s red cells carry one,
both or neither of the A and B antigens.
Normal healthy individuals, from early childhood, make red cell antibodies against the
A or B antigens that are not expressed on their own cells. These naturally occurring
antibodies are mainly IgM immunoglobulins and can rapidly bind to and destroy red
cells carrying the corresponding antigen.
If ABO incompatible red cells are transfused, intravascular haemolysis of these red
cells can occur. An ABO incompatible transfusion reaction may result in overwhelming
disruption of haemostatic equilibrium and complement activation, resulting in shock
and renal failure. This can be fatal, even after transfusion of a small volume of
incompatible blood.
Table 4.1: ABO Blood Groups and Antibodies
Patient’s ABO
Red Cell ABO
Plasma ABO
Blood Group
Antigens
Antibodies
A
A
Anti-B
B
B
Anti-A
AB
A and B
None
O
None
Anti-A and Anti-B
4.2
Avoiding ABO Incompatible Transfusions
Safe transfusion depends on avoiding ABO incompatibility between the patient and
transfused blood components, either between donor red cells and the patient’s ABO
antibodies or conversely donor plasma ABO antibodies and the patient’s red cells.
Page 34
Transfusion Medicine Handbook 3rd Edition
Red cells of the same ABO group as the patient, i.e., ABO identical, should normally
be selected. Only when these are unavailable should alternative ABO compatible red
cells be selected (see Table 4.9:
ABO Compatibility for Red Cell Components). In life-
threatening situations, where a confirmed blood group for the patient is not available,
group O red cells should be given. Group O donations identified as emergency units
have low levels of anti-A and anti-B to avoid potential sensitisation and destruction of
the patient’s red cells (in non-group O recipients).
Platelet concentrates should ideally be ABO identical or alternatively ABO compatible
with the patient’s red cells (see Table 4.11:
ABO Compatibility for Platelet Components).
This is however not a strict requirement and, due to logistics or supply issues, platelets
with a different ABO group may be supplied in clinically urgent situations following
consultation with an NZBS Transfusion Medicine Specialist. Additional pretransfusion
testing is not required for platelets.
Fresh frozen plasma and cryoprecipitate should be ABO compatible with
the patient’s red cells (see Table 4.14:
ABO Compatibility of Fresh Frozen Plasma
and Cryoprecipitate). Additional pretransfusion testing is not required for FFP and
cryoprecipitate.
4.3
RhD Antigen
After ABO, the RhD antigen ranks next in importance for transfusion. Patients and
blood donors are routinely typed for the RhD antigen and on the basis of its presence
or absence are called either RhD positive or RhD negative.
Antibodies to the RhD antigen only occur as a result of transfusion or pregnancy in
individuals who are RhD negative. The RhD antigen is highly immunogenic and a RhD
negative person only needs to be exposed to a small volume of RhD positive red cells
for immunisation to occur, stimulating production of anti-D.
Red cell and platelet transfusions are normally of the same RhD type as the patient.
RhD negative components may be given to RhD positive recipients without any risk
of immunisation.
In life-threatening situations or where RhD identical components are not readily
available, it may be necessary to transfuse RhD negative recipients with red cells or
platelets from a RhD positive donor. In these circumstances the blood bank will provide
guidance. It is essential that the treating clinician is informed and the appropriateness
of administration of prophylactic anti-D immunoglobulin considered. In the case of RhD
negative females with child-bearing potential, transfusion of RhD positive red cells must
only be considered following discussion with a NZBS Transfusion Medicine Specialist.
If supplies of RhD negative red cells are low, RhD positive red cells may be provided
by the blood bank for RhD negative males and for females beyond reproductive years.
Residual red cells in RhD positive platelet components may sensitise RhD negative
patients to form anti-D. When platelet components from a RhD positive donor are
transfused into a RhD negative recipient, in particular females of childbearing age or
female children, prophylactic anti-D immunoglobulin must be considered.
Section 5.4.7:
Rh(D) Immunoglobulin-VF contains guidelines on dosing of prophylactic
anti-D following transfusion of RhD positive blood components.
Transfusion Medicine Handbook 3rd Edition
Page 35
4.4
Other Blood Group Systems
Red cells possess many different antigens. Transfusion or pregnancy can stimulate
antibody production where a person is exposed to a red cell antigen that they lack.
Some of these antibodies can cause transfusion reactions or haemolytic disease of
the fetus and newborn (HDFN). Before transfusion and during pregnancy it is important
to detect clinically significant antibodies in a patient so that compatible red cells can
be provided and appropriate advice given during pregnancy.
4.5
Cytomegalovirus (CMV)
Prior to the introduction of routine prestorage leucodepletion, CMV was readily
transmitted by transfusion. CMV can cause severe and even fatal disease in certain
immunocompromised patients not previously exposed to the virus. Such patients
should receive CMV ‘safer’ blood components, i.e., components that have undergone
prestorage leucodepletion or found to be CMV-antibody negative.
The use of either prestorage leucodepletion or selection of CMV-antibody negative
blood components, obtained from a regular donor who has donated at least once in
the preceding 6 months, significantly reduces the risk of CMV transmission and CMV
disease in susceptible recipients. However, neither method alone or in combination
can completely avoid transmission from the occasional donor with CMV viraemia in
the “window” period prior to the development of antibodies following acute infection
or when reactivation of latent infection occurs.
Since all blood components in New Zealand are leucodepleted, NZBS has adopted
a policy that restricts the requirement for the use of blood components from CMV-
antibody negative donors to:
■
Intrauterine and neonatal transfusion.
■
A selected individual patient following discussion and agreement between the
treating clinician and a NZBS Transfusion Medicine Specialist.
4.6 Irradiation
Transfusion-associated graft-versus-host disease (TA-GVHD) is a rare but usually fatal
complication of transfusion. The risk associated with an individual transfusion depends
on the number and viability of contaminating lymphocytes, the susceptibility of the
patient’s immune system to donor lymphocyte engraftment (primarily related to the
degree of T-cell immunosuppression) and the degree of immunological incompatibility
between donor and patient.
Cellular components, with the exception of thawed cryopreserved haematopoietic
progenitor cells (HPC) intended for transplantation, must be irradiated to prevent
TA-GVHD in at-risk patients. Frozen plasma components and fractionated products
do not require irradiation.
The following tables summarise the clinical indications for blood component irradiation
recommended by the 2011 Australian & New Zealand Society of Blood Transfusion
(ANZSBT)
Guidelines for Prevention of Transfusion-Associated Graft-Versus-Host
Disease and the 2010 BCSH
Guidelines on the Use of Irradiated Blood Components.
Page 36
Transfusion Medicine Handbook 3rd Edition
Table 4.2: Definite Clinical Indications for Use of Irradiated Blood Components
Directed donations (from blood relatives)
HLA selected/matched platelet transfusions
Granulocyte transfusions
Intrauterine and all subsequent neonatal exchange transfusions
Congenital cellular immunodeficiency disorders
Allogeneic and autologous haematopoietic stem cell transplantation (HSCT)
Hodgkin lymphoma
Purine nucleoside analogues and the related agent bendamustine1 for malignant or
non-malignant disorders
Alemtuzumab for malignant or non-malignant disorders
1Bendamustine combines the alkylating properties of mechlorethamine and the purine antimetabolite
properties of benzimidazole.
Table 4.3: Possible Clinical Indications for Use of Irradiated Blood Components
Premature infants and infants weighing less than 1300g
All newborn infants
Acute leukaemia
Non-Hodgkin lymphoma
Patients with B-cell malignancy who receive non-nucleoside analogue containing
chemotherapy and/or radiotherapy leading to lymphopenia < 0.5 x109/L
T-cell malignancy
Patients receiving high doses of chemotherapy and/or irradiation sufficient to cause
lymphopenia < 0.5 x109/L
Patients receiving long term or high dose steroids as therapy for malignant disorders
Aplastic anaemia receiving immunosuppressive therapy
Massive transfusion for trauma
Table 4.4: No Clinical Indication for Use of Irradiated Blood Components
HIV/AIDS (where none of the above listed indications apply)
Congenital humoral deficiency disorders
Solid organ transplantation
Transfusion Medicine Handbook 3rd Edition
Page 37
Table 4.5: Recommendations on Duration of Use of Irradiated Blood Components
Autologous HSCT
From 7 days prior to initiation of conditioning and until at least three months post-
autograft, or six months if total body irradiation (TBI) is used.
Allogeneic HSCT
From initiation of conditioning and continued while post-transplant GVHD prophylaxis
is given, for a minimum of 6-12 months or until lymphocytes > 1x109/L. Patients
with active chronic post-transplant GVHD should continue to receive irradiated
components.
Aplastic anaemia treated with ATG immunosuppression
From initiation of therapy. No clear recommendations as to duration, but possibly until
lymphocytes > 1x109/L.
Hodgkin lymphoma
At all stages of disease and therapy. Continued indefinitely.
Purine nucleoside analogues1
and bendamustine
From initiation of therapy. Continued indefinitely.
Alemtuzumab
From initiation of therapy. No clear recommendations as to duration, but at least 12
months from last dose.
1Fludarabine, deoxycoformycin (pentostatin), chlorodeoxyadenosine (cladribine) and clofarabine.
Page 38
Transfusion Medicine Handbook 3rd Edition
esh is
e issued in this
educed by blood loss or
ease tissue oxygenation
ed cells ar
Indication for Use / Comments
irtually all r
Used to incr
when critically r
anaemia
V
form
As for Red Cells Resuspended
Not suitable for large volume transfusion
or exchange transfusion
Potassium content, when fr
2.0 mmol/L, and at expiry 15-35
mmol/L (may be raised if component is
irradiated)
Must be irradiated
Expiry
35 days
(2 - 6°C)
35 days
(2 - 6°C)
5 days
(2 – 6°C)
24 hrs once
irradiated
0.50 - 0.70
0.50 - 0.70
0.75 - 0.90
Haematocrit (%)
298
70
206
Typical Mean Volume (mL)
< 5
om NZBS
es of
es of
om whole
vailable from NZBS
ed fr
vailable fr
eplaced by additive
epar
ed to specified haematocrit
ed cell antibodies)
emoval of plasma
esh whole blood donation (
epar
Description
Red cells pr
blood donation with plasma
removed and r
solution
As for Red Cells Resuspended
Split into equal volume aliquots
suitable for neonatal use
CMV negative
Does not contain high titr
anti-A or anti-B
Fr
days old) collected into CPD and
pr
by r
CMV negative
Does not contain high titr
anti-A or anti-B (also negative for
other r
Blood Components A
4.7
Table 4.5: Red Cell Components A
Component
Red Cells
Resuspended
Red Cells
Resuspended
Neonatal
Red Cells For
IUT
Transfusion Medicine Handbook 3rd Edition
Page 39
n
oduct
cuit in
e the infant has
e indicated
educed by blood loss or
eceived IUT
ease tissue oxygenation
diac bypass cir
esponse to persistent allergic
ed cells ar
ement only wher
oduct of choice for neonatal exchange
eviously r
Indication for Use / Comments
Pr
transfusion
Exchange transfusion for clinically
significant anaemia with or without
hyperbilirubinaemia due to haemolytic
disease of the fetus and newbor
(HDFN) or other causes
Should be irradiated if for neonatal
exchange transfusion, although a strict
requir
pr
Can also be used for large volume
transfusions in neonates and for adult
transfusions
Used in r
reactions, for some IgA deficient
patients with anti-IgA, and when anti-T
depleted r
Restricted access to this pr
Used to incr
when critically r
anaemia
Primary car
paediatric bypass surgery
Expiry
5 days
(2 - 6°C)
for neonatal
exchange
transfusion
28 days
(2 - 6°C)
for other
uses
24 hrs
(2 - 6°C)
CPD-A1: 35
days
(2 - 6°C)
CPD: 28
days
(2 - 6°C)
0.45 - 0.55
0.40 - 0.70
0.30 - 0.50
Haematocrit (%)
360
328
493
Typical Mean Volume (mL)
ed to
epar
emoval
es of
otein
ected donations
vailable from NZBS continued
esidual plasma pr
e issued in this form
Description
Whole blood donation pr
specified haematocrit by r
of plasma
CMV negative
Does not contain high titr
anti-A or anti-B
Red cell donation washed leaving
total r
< 0.5 g per unit
Resuspended in additive solution
Whole blood donation collected
into CPD or CPD-A1
Autologous and dir
ar
ashed
Component
Whole Blood
Plasma
Reduced
Red Cells
W
Whole Blood
Table 4.5: Red Cell Components A
Page 40
Transfusion Medicine Handbook 3rd Edition
ded at the
om whom it
ed for
e deficiency of
oblem
A or HLA antibodies
esponse
ovides an adequate clinical
eased platelet consumption
ovide compatible platelets for
efractory to random donor
ith incr
Indication for Use / Comments
Replacement wher
platelets or platelet dysfunction is
causing or may cause a significant
haemostatic pr
One unit pr
response in most adult patients
W
or destruction the dose is determined
by the clinical r
As for Platelet Pool
Also to pr
patients with HP
and r
platelets
Indication for Use / Comments
Unit is labelled and stor
transfusion to the patient fr
was collected. If not transfused to the
identified patient, unit is discar
designated expiry of the component
Expiry
Expiry
7 days
(20 - 24°C
with constant
agitation)
If irradiated
original expiry
applies
As for Platelet
Pool
)9
(x 10
■≥240
≥240
Platelets
Haematocrit (%)
307
228
Typical Mean
Typical Mean Volume (mL)
Volume (mL)
esis
om the
vailable from NZBS continued
vailable from NZBS
AS)
om a single apher
AS)
ements the same as Whole
fy coats of four whole blood
Description
Platelet pool derived fr
buf
donations
Resuspended in platelet additive
solution (P
Platelets fr
donation
Resuspended in platelet additive
solution (P
Description
Specifications and storage
requir
Blood
esis
Component
Whole Blood
Autologous
Table 4.5: Red Cell Components A
Table: 4.6: Platelet Components A
Component
Platelet Pool
Platelets
Apher
Transfusion Medicine Handbook 3rd Edition
Page 41
eactions to
e allergic r
ent sever
Indication for Use / Comments
As for Platelet Pool
May be indicated in patients who have
recurr
plasma-containing components and
in some IgA deficient patients with
anti-IgA antibodies
Expiry
As for Platelet
Pool
Closed
system:
24 hrs
(20 - 24°C
with constant
agitation)
Open system:
6 hrs
(20 - 24°C
with constant
agitation)
)9
(x 10
≥40
■≥240
Platelets
45
227
Typical Mean
Volume (mL)
esis
otein
vailable from NZBS continued
esis donation
om a single apher
esidual plasma pr
oved platelet additive
Description
Single apher
suspended in plasma
Divided into equal volume aliquots
suitable for neonates
CMV negative
Platelets fr
donation, washed with an
appr
solution.
Total r
< 0.5 g per unit.
esis
esis
ashed
Component
Platelets
Neonatal
Apher
Platelets
Apher
W
Table: 4.6: Platelet Components A
Page 42
Transfusion Medicine Handbook 3rd Edition
e is
■
e ther
ed to
d adult
openia
esponding
ecovery of
equir
/L, bone
fy coats
9
Comments
ow hypoplasia and
ow function
esis granulocytes
Indication for Use /
ofound neutr
Supportive therapy for
pr
<0.5 x 10 marr infection not r to antibiotics within 48 hours, and wher potential for r marr
Irradiate prior to use
Clinical indications as for
apher
Multiple buf
≥10 will be r achieve a standar therapeutic
esis
Expiry
24 hrs
(20 - 24°C)
As for
Granulocyte
Apher
)9
(x 10
≥ 200
≥ 50
Platelets
)9
(x 10
≥10
≥ 1
Granulocytes
50
Variable
Typical Mean Volume (mL)
vailable from NZBS
om
om a
esis
ed cells and
ed cells
Description
Granulocytes (with
some r
platelets) fr
single apher
donation
CMV negative if
indicated
Granulocytes (with
some r
and platelets) fr
single whole blood
donation
esis
fy Coat
Table 4.7: Granulocyte Components A
Component
Granulocyte
Apher
Buf
Transfusion Medicine Handbook 3rd Edition
Page 43
educed
ozen
oteins when
e critically r
esh Fr
Indication for Use/Comments
Replacement of coagulation factors
and other plasma pr
levels or activity ar
As for Plasma Fr
eeze
efr
ozen
esh Fr
Expiry
24 months
(-25°C or below)
24 hrs post
thawing
(2 - 6°C)
Do not r
As for Plasma
Fr
(g)
Mean
Fibrinogen
Factor VIIIc
≥ 0.7
(IU/mL)
≥ 0.7
279
60
vailable from NZBS
Typical Mean Volume (mL)
esis
om
e fr
ozen
es of anti-A
ocedur
esh Fr
Description
Plasma collected
using an apher
pr
a male donor (to
reduce the risk
of TRALI), rapidly
frozen within 8
hours of collection
to maintain labile
coagulation
factors
As for Plasma
Fr
Split into equal
volume aliquots
suitable for
neonates
Does not contain
high titr
or anti-B
esh
esh
ozen
ozen Neonatal
Table 4.8: Frozen Plasma Components A
Component
Plasma Fr
Fr
Plasma Fr
Fr
Page 44
Transfusion Medicine Handbook 3rd Edition
eating
illebrand
eatment of
ombocytopenic
eat von W
opriate or unavailable can
ombotic Thr
ect haemostatic defects
Indication for Use/Comments
Used for plasma exchanges
particularly in the tr
Thr
Purpura (TTP)
Corr
associated with fibrinogen deficiency
and dysfibrinogenaemia
May be of clinical value for tr
bleeding due to uraemia
If specific concentrate therapy is
inappr
be used to tr
disease, haemophilia A or factor XIII
deficiency
oom
e)
eeze
ozen
efr
ed at r
esh Fr
Expiry
As for Plasma
Fr
24 months
(-25°C or below)
4 hrs post thawing
(stor
temperatur
Do not r
(g)
1.2
Mean
Fibrinogen
Factor VIIIc (IU/mL)
≥ 1.5
473
98
Typical Mean Volume (mL)
vailable from NZBS continued
,
,
esis
esis
om
esh
e fr
natant
ecipitate
ce of
ozen depleted
illebrand factor
onectin
ocedur
illebrand factor
onectin
Description
Super
from apher
cryopr
frozen within 2
hours of collection
Plasma Fr
Fr
of fibrinogen,
factor VIII, von
W
factor XIII, and
fibr
Plasma collected
using an apher
pr
a male donor (to
reduce the risk of
TRALI)
Concentrated
sour
fibrinogen,
factor VIII, von
W
factor XIII, and
fibr
esis
ecipitate
esis - High
Component
Plasma
Cryodepleted
Apher
Cryopr
Apher
Fibrinogen
Table 4.8: Frozen Plasma Components A
Transfusion Medicine Handbook 3rd Edition
Page 45
4.8
Red Cell Components
ABO Compatibility
It is important that transfusion recipients receive red cell components compatible with
their own ABO group. Incompatible transfusion can result in serious harm or death
of the recipient.
It is best practice to transfuse donor red cells that are matched for the recipient. In some
circumstances it may not be possible to transfuse the recipient with donor cells of the
same group. The following table outlines alternative donor groups that may be given to
a recipient if supplies of ABO identical red cells are not available or are in short supply.
Table 4.9: ABO Compatibility for Red Cell Components
Recipient
Compatible Donor Groups
Group
(in order of preference)
Unknown
Group O§ cells
Preferably RhD negative for premenopausal females
The transfusion of group O cells is usually continued only until the
patient’s blood group is known
O
O
A
A or O
B
B or O
AB
AB or A or B or O§
§These group O components should test negative for ‘high titre’ anti-A and anti-B.
RhD Compatibility
Red cell and platelet transfusions are normally of the same RhD type as the patient.
RhD negative components may be given to RhD positive recipients without any risk
of immunisation.
In life-threatening situations it may be necessary to transfuse RhD negative recipients
with RhD positive red cells. In these circumstances the blood bank will provide
guidance. It is essential that the treating clinician is informed and the appropriateness
of administration of prophylactic anti-D immunoglobulin considered.
If supplies of RhD negative red cells are low, RhD positive red cells may be provided
by the blood bank for RhD negative males and for females beyond reproductive years.
In the case of RhD negative females with child-bearing potential, transfusion of RhD
positive red cells must only be considered following discussion with a NZBS Transfusion
Medicine Specialist.
Section 5.4.7:
Rh(D) Immunoglobulin-VF contains guidelines on dosing of prophylactic
anti-D following transfusion of RhD positive components.
Guidelines for Appropriate Use
In deciding whether or not to transfuse red blood cells, the haemoglobin level, although
important, should not be the sole deciding factor. Patient factors, signs and symptoms
Page 46
Transfusion Medicine Handbook 3rd Edition
of hypoxia, ongoing blood loss, the degree of urgency required in correcting the
anaemia and the risk of transfusion-related adverse effects should all be considered.
The key issue is delivery of adequate amounts of oxygen to tissues. For a 70-80
kg patient, a transfusion of 4-5 mL/kg will increase the circulating haemoglobin by
about 10 g/L.
Some specific factors to consider before deciding to transfuse:
■
Cardiopulmonary reserve
If cardiac or pulmonary function is not normal, it may be necessary to consider
transfusing at a higher haemoglobin level.
■
Volume of blood loss
Clinical assessment should attempt to quantify the volume of blood loss before,
during, and after surgery, to ensure maintenance of adequate blood volume.
■
Oxygen consumption
This may be affected by a number of factors including fever, anaesthesia,
shivering and thyrotoxicosis. If oxygen consumption is increased it may be
necessary to consider transfusing at a higher haemoglobin level.
■
Atherosclerotic disease Critical arterial stenosis to major organs, particularly the heart, may modify
indications for the use of red cells.
The Australian National Blood Authority
(NBA) Patient Blood Management Guidelines
and the 2012 British Committee for Standards in Haematology (BCSH)
Guidelines on
the Management of Anaemia and Red Cell Transfusion in Adult Critically Ill Patients
provide the following recommendations for red cell component use in adults.
Table 4.10: Indications for Transfusion of Red Cells in Relation to Haemoglobin Level
Hb Level
Indication
< 70 g/L
Transfusion of red cells is usually indicated however a lower
threshold may be acceptable in patients without symptoms and
where specific therapy is available, e.g., cobalamin or iron deficiency
anaemia.
< 80 g/L
Transfusion of red cells is likely to be appropriate in patients
with acute coronary syndrome and the haemoglobin should be
maintained > 80-90 g/L.
70 - 100 g/L
Transfusion of red cells is likely to be appropriate during surgery
associated with major blood loss or with evidence of impaired tissue
oxygen delivery.
> 90 g/L
Transfusion of red cells is not likely to be appropriate in the critically
ill, in whom a restrictive transfusion policy is associated with
reduced mortality. Exceptions may include patients with any of the
following: sepsis together with evidence of impaired tissue oxygen
delivery, subarachnoid haemorrhage, ischaemic stroke, and cerebral
ischaemia complicating traumatic brain injury.
> 100 g/L
Transfusion of red cells is not likely to be appropriate unless there
are specific indications.
Transfusion Medicine Handbook 3rd Edition
Page 47
Restrictive Red Cell Transfusion Policy
The decision to transfuse should take into account the risks, benefits and alternatives
available, and should not be based on haemoglobin level alone. With this in mind,
increasingly restrictive transfusion policies are being implemented in a variety of
clinical settings.
Policies aim to safely provide the minimum amount of blood required for an individual
patient through setting appropriate transfusion triggers and, where indicated,
encouraging assessment of the patient’s clinical status after transfusion of each single
unit of red blood cells before determining that further transfusion is required. Restrictive
policies are supported by evidence from a number of clinical areas of improved or
equivalent clinical outcomes after minimising exposure to allogeneic blood.
One such policy, the 2014 Australian NBA Single Unit Transfusion Guide (available
at http://www.blood.gov.au/single-unit-transfusion), has been designed for use in
stable, normovolaemic adult patients, in an inpatient setting, who do not have clinically
significant bleeding.
4.9
Platelet Components
ABO and RhD Compatibility
Transfused platelets should ideally be the same ABO and RhD type as the recipient
although platelet stocks may not always permit this. Preferably platelet components
that are ABO/RhD compatible but plasma incompatible should be selected for
transfusion. This compromise is relatively free of adverse reactions although antibodies
in the plasma may occasionally cause a haemolytic reaction or a transiently positive
direct antiglobulin test (DAT).
Table 4.11: ABO Compatibility for Platelet Components#
Recipient
Compatible Donor Groups
Group
(in order of preference)
Unknown
Group O§ or A platelets
Preferably from RhD negative donor for premenopausal females
O
O or A
A
A or O§
B
B or O§
AB
AB or A or B or O§
#NZBS routinely makes platelet components from group A and O donors only.
§These group O components should test negative for ‘high titre’ anti-A and anti-B.
In the event of life-threatening bleeding, ABO and/or RhD incompatible platelet
components may be transfused.
Rh antigens are not expressed on platelets and RhD incompatibility has no effect
on the survival of transfused platelets. Residual red cells in RhD positive platelet
components may however sensitise RhD negative patients to form anti-D. When
platelet components from a RhD positive donor are transfused into a RhD negative
Page 48
Transfusion Medicine Handbook 3rd Edition
recipient, and in particular females of childbearing age or female children, administration
of prophylactic anti-D immunoglobulin must be considered. Section 5.4.7:
Rh(D)
Immunoglobulin-VF contains guidelines on dosing of prophylactic anti-D following
transfusion of RhD positive blood components.
Clinical Indications
The 2006 BCSH
Guidelines on the Management of Massive Blood Loss and the
Australian NBA
Patient Blood Management Guidelines along with expert consensus
opinion provide the following recommendations for platelet component use in adults.
Table 4.12: Clinical Indications for Use of Platelets
Indication
Management
Chemotherapy-induced ■ Prophylactic platelets are indicated in patients when
Bone Marrow Failure
the platelet count is <10 x 109/L in the absence of
risk factors.
■
Prophylactic platelets are indicated in patients when
the platelet count is <20 x 109/L in the presence of
risk factors for systemic haemostatic failure such as
fever or minor bleeding.
Surgery/Invasive
■
Patients with a platelet count ≥50 x 109/L can
Procedure
generally undergo invasive procedures without
serious bleeding; lower counts may be tolerated in
certain clinical situations.
■
For surgical procedures with high risk of bleeding
(e.g., ocular or neurosurgery) it may be appropriate
to maintain the platelet count ≥100 x 109/L.
Platelet Function
■
Rarely require platelet transfusion. May be
Disorders
appropriate in inherited or acquired disorders.
■
The platelet count is not a reliable indicator of platelet
haemostatic function (e.g., following extended cardiac
bypass surgery of more than 2 hours duration or
administration of anti-platelet medications).
■
Alloimmunisation to missing glycoproteins may occur
if platelets are given to patients with certain inherited
functional defects.
Bleeding
■
May be appropriate in any patient in whom
thrombocytopenia is considered to be a major
contributing factor.
■
In critically ill patients, in the absence of bleeding,
prophylactic platelet transfusion to prevent bleeding
may be appropriate at a platelet count <20 x 109/L.
■
In non-critically ill medical patients, lower platelet
counts may be tolerated.
■
In patients with chronic marrow failure syndromes,
prophylactic platelet transfusions may lead
to alloimmunisation and subsequent platelet
refractoriness.
Transfusion Medicine Handbook 3rd Edition
Page 49
Table 4.12: Clinical Indications for Use of Platelets continued
Indication
Management
Massive Haemorrhage/
■
To adequately maintain the platelet count >50 x 109/L
Massive Transfusion
while allowing for a margin of error, a transfusion
trigger of 75 x 109/L is recommended.
■
The platelet count should be maintained >100 x 109/L
in the presence of diffuse microvascular bleeding,
multiple or CNS trauma, or platelet dysfunction.
■
Massive transfusion protocols, recognising that a
platelet count <50 x 109/L can be expected after
replacement of 2 x blood volume, empirically include
platelet concentrates. Regular laboratory and/or
point-of-care monitoring may identify patients likely
to benefit from additional platelet transfusion.
Patients Refractory to Platelet Transfusion
A proportion of patients become refractory to random platelet transfusions. When
a platelet transfusion fails to achieve the desired response it is important to find
out whether the failure is due to rapid immunological or non-immunological platelet
consumption. Clinical factors such as sepsis, disseminated intravascular coagulation
(DIC), and splenomegaly are more common than alloimmunisation as the cause of
platelet refractoriness.
Identifying patients with antibodies to human leucocyte antigens (HLA) or human
platelet antigens (HPA) is important since the use of HLA- or HPA-matched platelet
components may result in improved transfusion response.
In deciding how to treat a refractory patient there may be a number of appropriate
strategies for improving the response to platelet transfusions such as matching for
HLA or HPA, increasing the transfused dose or even discontinuing transfusion.
NZBS has produced
Guidelines for the Management of Patients Refractory to
Platelets
from which the table below is taken. These guidelines can be found in the
Clinical
Information section of the NZBS website (www.nzblood.co.nz).
Table 4.13: Options for Managing Patients Refractory to Platelets
Indication
Management
Patient’s HLA type not
■
Consider transfusing ABO compatible, single
known and serum sample(s)
donor platelets (preferably ‘double-dose’).
not yet available
Patient serum samples
■
Platelets from the donation inventory can be
available but HLA type not
prospectively crossmatched and compatible
known
units selected for transfusion, although this is
not the preferred option.
Patient’s HLA type is known, ■ Consider transfusing HLA-matched platelets
and HLA-matched platelets
prior to the completion of antibody testing, as
are available
HLA immunisation is the most common cause
of immune refractoriness.
Page 50
Transfusion Medicine Handbook 3rd Edition
Table 4.13: Options for Managing Patients Refractory to Platelets continued
The patient has HLA and/or
■
Antigen negative (compatible) platelets should
HPA antibodies
be selected for transfusion.
■
Further antibody testing is recommended every
3 months or if refractoriness returns.
The patient does not have
■
Consideration should be given to non-
HLA or HPA antibodies
immunological causes for which a
haematologist or NZBS TMS/MO will advise
management options.
Matched platelets are prepared for a specific recipient after consultation with a NZBS
Transfusion Medicine Specialist. They normally require at least 48 hours notice, as, in
addition to usual pre-release testing for bacterial contamination, suitable donors first
need to be identified and then bled.
Contraindications
Transfusion of platelets is generally contraindicated in the following conditions:
■
Thrombotic thrombocytopenic purpura (TTP)
■
Haemolytic uraemic syndrome (HUS)
■
Heparin-induced thrombocytopenia (HIT)
■
Posttransfusion purpura (PTP)
Severe adverse reactions have been reported in patients with TTP and HIT following
platelet transfusion. Platelet transfusion to these patients may also precipitate
thrombotic events and can aggravate their clinical condition.
In this next group, platelet transfusion is unlikely to cause any sustained increase of
platelet count:
■
Immune thrombocytopenic purpura (ITP)
■
Drug-induced thrombocytopenia of immune origin
Prophylactic use of platelet transfusion in these patients is of little benefit but
platelet transfusion may be useful to stop active bleeding. These complex disorders
of haemostasis should be managed in consultation with, or supervised by, a
haematologist.
Adverse Reactions
Adverse reactions to platelets are predominantly allergic or febrile non-haemolytic
transfusion reactions. The frequency of adverse reactions during transfusion of platelet
concentrates has reduced following the introduction of platelet additive solution (PAS)
as suspension medium.
4.10 Granulocyte Components
Granulocyte components may by collected by apheresis or harvested from buffy coats
obtained from whole blood donations. Granulocytes must be ABO and RhD compatible
with the recipient and must be irradiated before transfusion.
Transfusion Medicine Handbook 3rd Edition
Page 51
NZBS has produced a
Policy for Collection and Transfusion of Granulocytes available
in the
Clinical Information section of the NZBS website (www.nzblood.co.nz).
A request for granulocyte components must be made in consultation with a NZBS
Transfusion Medicine Specialist.
Clinical Indications
The literature has shown granulocyte transfusions may be beneficial and even life-saving
in severely neutropenic patients with systemic bacterial or invasive fungal infections
not responding to antimicrobial therapy after 24 to 48 hours, and in whom there is
the potential for recovery of marrow function.
The literature on prophylactic granulocyte transfusions does offer some support for
high-dose crossmatch compatible granulocyte transfusions. The Council of Europe
does not currently include prophylaxis as an indication for granulocyte transfusions.
Adverse Reactions
As with other blood components, adverse reactions may occur, with febrile non-
haemolytic transfusion reactions (FNHTR) being the most common and often dose
related. The development of HLA antibodies and subsequent immune refractoriness
to platelet components may further complicate blood transfusion support in recipients
of granulocytes.
4.11 Plasma Components
ABO Compatibility
To avoid red cell haemolysis caused by transfusion of donor anti-A or anti-B, the
ABO group of plasma components should be compatible with the ABO group of
the recipient.
If high titre anti-A or anti-B are present in the plasma, the unit will be labelled accordingly
and should only be transfused to a recipient with a compatible ABO group.
Table 4.14: ABO Compatibility of Fresh Frozen Plasma and Cryoprecipitate#
Patient’s ABO Blood Group
ABO Group of Plasma Component
ABO Unknown
AB if urgent
O
O§ or A or B or AB
A
A or AB
B
B or AB
AB
AB (A if AB is unavailable)
# Cryoprecipitate is free from high titre isoagglutinins anti-A and anti-B and can usually be given regardless
of ABO group, however it is usual to follow the compatibility rules.
§ Group O fresh frozen plasma is no longer routinely available.
Page 52
Transfusion Medicine Handbook 3rd Edition
RhD Compatibility
Although frozen plasma components may contain small amounts of red cell stroma,
sensitisation following transfusion of RhD positive units is most unlikely, as stroma
is less immunogenic than intact red cells. Therefore, FFP and cryoprecipitate of any
RhD type may be given, regardless of the RhD type of the recipient. No prophylactic
anti-D immunoglobulin need be given if RhD negative patients receive RhD positive
FFP or cryoprecipitate.
Clinical Indications
Guidance on the indications for and dose of plasma components may be sought from
a haematologist or NZBS Transfusion Medicine Specialist.
The following references provide recommendations for the appropriate use of plasma
components:
■
Australian NBA
Patient Blood Management Guidelines
■
2013 ASTH
Consensus Guidelines for Warfarin Reversal
■
2004 BCSH
Guidelines for the Use of Fresh-Frozen Plasma, Cryoprecipitate
and Cryosupernatant
Adverse Reactions
Acute transfusion reactions to plasma components may be seen and include febrile
non-haemolytic transfusion reactions (FNHTR), allergic reactions to plasma protein
antigens, circulatory overload (TACO) following transfusion of volumes excessive for the
patient, haemolytic reactions due to ABO incompatible plasma, bacterial contamination
and transfusion-related acute lung injury (TRALI), although the latter has reduced in
frequency following the introduction of male-only plasma (see Chapter 7:
Adverse
Effects of Transfusion).
Contraindications
It is not appropriate to use plasma components as a plasma expander or for
replacement of plasma proteins in chronic hypoproteinaemic states. Fresh frozen
plasma is generally not used for plasma exchange procedures except where performed
for thrombotic thrombocytopenic purpura or in the presence of severe coagulopathy.
The use of fresh frozen plasma is generally not considered appropriate for treating
immunodeficiency states or for urgent reversal of vitamin K deficiency due to warfarin
anticoagulation when a prothrombin complex concentrate (PCC) is readily available.
4.12 Fresh Frozen Plasma
Fresh frozen plasma is collected using an apheresis procedure and rapidly frozen
within 8 hours of collection to maintain labile coagulation factors.
Transfusion Medicine Handbook 3rd Edition
Page 53
Table 4.15: Clinical Indications for Use of FFP
Indication
Comments
Single Coagulation
■
When a specific factor concentrate is unavailable or is
Factor/Protein
inappropriate, including isolated deficiency of factor II,
Deficiencies
V, VII, X, XI, XIII, antithrombin, C1-esterase inhibitor or
pseudocholinesterase.
■
The dose will depend on the clinical circumstances
and the required level of activity in the patient. Consult
a haematologist or NZBS TMS/MO.
Massive Blood
■
In patients with clinical y abnormal haemostasis and
Transfusion
reduced levels of coagulation factors following rapid
transfusion of large volumes of blood.
■
Massive transfusion protocols (MTP) empirically provide
FFP units in a ratio to red cells of 1:1.
■
Where a MTP is not available, an adult dose of 12-15
mL/kg (i.e., 1 L or 4 units FFP) is likely to be required
after 1-1.5 x blood volume replacement in order to
maintain the PT/APTT <1.5 x mean control.
■
In patients with ongoing blood loss and consumption of
coagulation factors, close monitoring of clinical status
and levels of coagulation factors should be used to
guide additional doses of FFP. Note however that the
INR/APTT may not fully correct with the use of FFP.
Reversal of
■
Use of FFP in this setting should be based on ASTH
Warfarin Effect
consensus guidelines (see Section 6.5.1:
Warfarin).
■
In summary, FFP should only be used to reverse
warfarin anticoagulation in the presence of bleeding
or prior to emergency surgery where a prothrombin
complex concentrate (PCC) is unavailable or deemed
inappropriate. In the case of life threatening bleeding,
FFP may have a supplementary role in addition to PCC
for warfarin reversal.
Thrombotic
■
Accepted treatment is plasma exchange using FFP or
Thrombocytopenic
cryosupernatant plasma as replacement fluid.
Purpura (TTP)
■
If apheresis is not immediately available, infusion of FFP
or cryosupernatant plasma may be given until plasma
exchange can be initiated.
Page 54
Transfusion Medicine Handbook 3rd Edition
Table 4.15: Clinical Indications for Use of FFP continued
Indication
Comments
Liver Disease
■
FFP may be appropriate in the presence of bleeding
and abnormal coagulation, or as prophylaxis prior to
invasive procedures.
■
Due to reduced levels of natural anticoagulant proteins
in liver disease, values of PT/APTT may overestimate
the bleeding risk in liver disease.
■
The usual adult dose is 10-15 mL/kg, however the
response in liver disease is variable, partial and transient
and close laboratory monitoring may be required.
Cardiac Surgery
■
There is no evidence that prophylactic use of FFP (or
cryoprecipitate and platelets) reduces bleeding.
■
FFP (and other components) may be used to correct
coagulopathy in bleeding patients, guided by clinical
response and laboratory monitoring.
Disseminated
■
Indicated only when the consumptive coagulopathy is
Intravascular
complicated by bleeding, in which case maintaining PT/
Coagulation (DIC)
APTT <1.5 x mean control and fibrinogen >1.5 g/L may
be of benefit.
■
The initial recommended dose is 15 mL/kg, however
this will depend on the severity of the DIC.
■
Subsequent doses should be guided by frequent
laboratory testing. Cryoprecipitate may be required if
fibrinogen is severely reduced.
4.13 Cryoprecipitate Apheresis - High Fibrinogen
Cryoprecipitate is a concentrated source of fibrinogen and also contains von Willebrand
factor, factor VIII, factor XIII and fibronectin.
A dose of one unit per 30 kg body weight will produce an increment in plasma fibrinogen
of approximately 1.0 g/L.
Table 4.16: Clinical Indications for Use of Cryoprecipitate
Indication
Comments
Disseminated
■
Consumptive fibrinogen deficiency is commonly
Intravascular
encountered in DIC.
Coagulation (DIC) with
Bleeding
■
At fibrinogen levels lower than 1.5 g/L and where
there is clinical bleeding, use of cryoprecipitate to
maintain the fibrinogen level above 1.5 g/L may be
indicated.
Transfusion Medicine Handbook 3rd Edition
Page 55
Table 4.16: Clinical Indications for Use of Cryoprecipitate continued
Indication
Comments
Fibrinogen Deficiency
■
Cryoprecipitate may be appropriate where there
and Dysfibrinogenaemia
is clinical bleeding in the event of an invasive
procedure, trauma or DIC.
■
The actual dose should be determined from the
recipient’s measured functional fibrinogen level, the
nature and degree of bleeding and other relevant
clinical factors.
Coagulation Factor
■
May be used as an alternative product for the
Deficiencies
treatment of bleeding associated with von
Willebrand disease, haemophilia A and deficiency
of factor XIII (fibrin stabilising factor) if specific
concentrates are not available or are considered
inappropriate.
■
The dose will depend on factor levels and the
nature and extent of bleeding.
Bleeding Associated
■
The transfusion of one unit per 30 kg body weight
with Uraemia
usually is effective in controlling bleeding however
repeated doses may be necessary.
4.14 Cryosupernatant Plasma
Cryosupernatant plasma is the supernatant from apheresis cryoprecipitate frozen within
2 hours of collection and is essentially FFP that has been depleted of factor VIII, von
Willebrand factor (high molecular weight multimers being more thoroughly removed
than smaller multimers), factor XIII, fibrinogen and fibronectin.
Cryosupernatant may be used as an alternative to FFP as a source of ADAMTS-13
protease (a disintegrin and metalloproteinase with a thrombospondin type 1 motif,
member 13) without replacing high molecular weight multimer vWF during plasma
exchange in the treatment of thrombotic thrombocytopenic purpura.
Page 56
Transfusion Medicine Handbook 3rd Edition
5
FRACTIONATED PRODUCTS
Fractionated products, also known as plasma derivatives, are partial y purified
therapeutic preparations of human plasma proteins. They are manufactured under
pharmaceutical conditions from large volumes of donor plasma with the final products
supplied as freeze dried powders or solutions.
Careful screening of every donation contributing to a plasma pool is vital since any
one donation could potentially introduce infectious agents into the pool. Even with
rigorous screening of every donor and infectious diseases testing of every donation,
some infectious agents might still find their way into the plasma pool. To counter this
risk, manufacturing processes include two or more specific steps to inactivate any
such agents that may escape detection.
Some points to note regarding transfusion of fractionated products:
■
The manufacturer’s instructions should be carefully read. Specific information
about the administration of each product is given in the product information
sheet, which comes packaged with each unit.
■
Products should be transfused promptly following issue from the blood bank. If
there is any delay they must be stored in a refrigerator at 2-8°C (unless otherwise
indicated by the manufacturer).
■
Freeze-dried preparations must be infused immediately after reconstitution. Do
not refrigerate after reconstitution.
■
Products not containing an antimicrobial preservative must be transfused within
3-4 hours of breaking the product seal.
■
Multiple vials or bottles of the same product may be pooled together immediately
prior to infusion to the patient.
■
Products should not be used if turbidity or particulate matter is present in the
vial or bottle. If observed this should be reported to NZBS.
■
ABO compatibility does not normally need to be considered. Residual anti-A and
anti-B in the final product are usually at clinically insignificant levels. However
in some situations, such as where high doses of Intragam® P are being given
to non-group O patients, these patients should be monitored for signs of
intravascular haemolysis.
■
Patients may experience adverse reactions due to transfusion of fractionated
products. These should be reported to NZBS using
a Notification of Adverse
Reaction to Fractionated Blood Product form.
The information contained within this chapter relates to the fractionated products
distributed by NZBS and has been adapted from the most recently available datasheet
and/or the manufacturer’s product information sheet for each product. These sources
should be consulted prior to administration. Current versions of the datasheets
can be accessed through the
Clinical Information section of the NZBS website
(www.nzblood.co.nz) and also the Medsafe website (www.medsafe.govt.nz). NZBS
Transfusion Medicine Specialists/Medical Officers and Transfusion Nurse Specialists
can also provide valuable information regarding the use of fractionated products.
Transfusion Medicine Handbook 3rd Edition
Page 57
5.1
Coagulation Factors
The National Haemophilia Management Group has produced guidelines for the
management of haemophilia, including vWD, and these are available from the
Haemophilia Centres. Most patients diagnosed with haemophilia A and B have their
treatment supervised by a specialist haematologist. For any individual patient, the
various fractionated and recombinant products used in the management of haemophilia
are not interchangeable without prior discussion with the treating haematologist.
The following guidelines are to assist in the immediate management of a patient until
consultation with a specialist haematologist or NZBS Transfusion Medicine Specialist.
The exact loading and maintenance dose and dosing interval should be based on the
patient’s clinical condition and response to therapy. Where possible, pre- and post-
infusion factor assays should be carried out, at least for the first course of treatment.
Coagulation factor therapy as a continuous infusion to cover surgical procedures should
be administered under the supervision of a specialist haematologist and with laboratory
tests performed to ensure that the desired plasma factor concentrations are achieved.
5.1.1 Biostate® (Factor VIII)
Biostate® is a high purity, sterile, freeze-dried powder containing purified human
coagulation factor VIII (FVIII) and human von Willebrand factor (vWF) complex. Biostate®
is available as vials of 500 IU and 1,000 IU factor VIII. The reconstituted product has a
vWF to FVIII ratio of 2:1 as detailed in Table 5.1. Biostate® contains 10 mg/mL human
albumin as a stabiliser.
The FVIII/vWF complex in Biostate® is purified from cryoprecipitate using selective
precipitation and chromatography steps. The manufacturing process of Biostate®
includes solvent detergent and dry heat treatment steps to reduce the potential for
viral transmission. The combination of solvent detergent, dry heat treatment, and
partitioning steps are effective for inactivation/removal of HIV, hepatitis A, hepatitis B,
and hepatitis C and also have some effect on parvovirus B19.
Table 5.1: Biostate® Composition
Active ingredients
Biostate®
IU/vial (nominal)
500 IU (50 IU/mL)
1000 IU (100 IU/mL)
Factor VIII
500
1000
vWF:RCo1
1000
2000
Reconstitution
10
10
volume (mL)
1vWF:Ristocetin Cofactor – an in vitro indicator of vWF activity.
Indications for Use
Biostate® is indicated for:
■
Treatment and prophylaxis of bleeding associated with FVIII deficiency due to
haemophilia A
Page 58
Transfusion Medicine Handbook 3rd Edition
■
Treatment and prophylaxis of bleeding associated with von Willebrand
disease (vWD) when desmopressin (DDAVP) treatment alone is ineffective or
contraindicated
The number of vials of Biostate® to be reconstituted for administration is determined by
dividing the total number of International Units (IU) required by 500 or 1,000 (according
to vial size being used), rounded up to the nearest whole number of vials.
Table 5.2: Biostate® Dosage Guidelines for Haemophilia A
Indication
Dose
Dose
Treatment
Target FVIII
(IU/kg)
Frequency
Duration
(%) (IU/dL)
(per day)
(days)
Minor haemorrhage
10-15
1-2
1-2
peak 20-30
Moderate to severe
haemorrhage
■ Haemarthroses
15-40
1-3
1-4
peak 30-80
Life threatening
haemorrhage
■ Intracranial
50-60
2-3
7-10
peak ≥100
haemorrhage
trough 80-100
Minor surgery
■ Loading dose
20-30
stat
preoperatively
peak 40-60
■ Maintenance1
15-30
1-2
≥4
trough 20-50
Major surgery
■ Loading dose
40-50
stat
preoperatively
peak 80-100
■ Maintenance1
10-25
1-3
≥7
trough 40-80
Dentistry2
■ Loading dose
35-40
stat
preoperatively
peak 70-80
■ Maintenance1
25-30
2
1-3
trough 50-60
Prophylaxis
25-40
3x/week
ongoing
trough 1
1An alternative is to use continuous infusion.
2 For single tooth extraction, extensive dental clearance or surgery, higher levels may be necessary for
longer periods of at least 6 - 10 days. The use of an antifibrinolytic agent such as tranexamic acid is
strongly recommended.
Transfusion Medicine Handbook 3rd Edition
Page 59
Table 5.3: Biostate® Dosage Guidelines for Von Willebrand Disease1
Indication
Dose (IU/kg)
Dose
Treatment Target FVIII/
Frequency Duration
vWF (%)
(per day)
(days)
(IU/dL)
FVIII:C vWF:RCo
Spontaneous 12.5-25
25-50
initial
-
peak vWF >50,
haemorrhage
FVIII >30
12.5
25
1-2
2-4
trough vWF >30
Minor surgery2
30
60
1
2-4
trough
vWF/FVIII >30
Major surgery2
30-40
60-80
preoperatively
-
peak vWF >100,
FVIII >60
15-30
30-60
1-2
5-10
trough
vWF/FVIII >50
Prophylaxis
12.5-25
25-40
3x/week
ongoing
trough 1
1 Dosage guidelines above are for patients with severely reduced vWF levels, e.g. <
10% of normal. Doses
may need to be adjusted down in patients with less severe vWF deficiencies (>
20% of normal) to ensure
that the desired plasma concentrations of vWF and FVIII are achieved. It is recommended that plasma
vWF and FVIII concentrations are determined at suitable time intervals.
2An alternative is to use continuous infusion.
Precautions
■
Allergic reactions
Allergic reactions or fever are rarely observed. Depending on the nature of an
adverse reaction, the rate of injection should be slowed or stopped to alleviate
symptoms.
■
Antibodies to factor VIII
Patients with congenital factor VIII deficiency may develop neutralising
alloantibodies (inhibitors) to factor VIII after treatment. If this occurs specialist
advice must be sought.
■
Antibodies to von Willebrand factor
Patients with von Willebrand disease, especially type 3 patients, may very
rarely develop neutralising alloantibodies (inhibitors) to von Willebrand factor.
Such antibodies may occur in close association with anaphylactic reactions.
Therefore patients experiencing anaphylactic reactions should be evaluated for
the presence of an inhibitor.
■
Thrombosis
Thromboembolic events have rarely been reported in vWD patients receiving
coagulation factor replacement therapy, especially in the setting of known risk
factors for thrombosis, and may be related to the generation of supranormal
FVIII levels.
5.1.2 MonoFIX®-VF (Factor IX)
MonoFIX®-VF is a sterile, freeze-dried powder containing purified human coagulation
factor IX. MonoFIX®-VF is available as vials of 500 IU and 1,000 IU factor IX. Each vial
also contains small amounts of heparin, antithrombin III and plasma proteins.
Page 60
Transfusion Medicine Handbook 3rd Edition
The factor IX in MonoFIX®-VF is purified using ion-exchange and heparin affinity
chromatography to remove other vitamin K-dependent factors II, VII and X. The
manufacturing process of MonoFIX®-VF includes solvent detergent treatment and
nanofiltration steps to reduce the potential for viral transmission. The current procedures
are effective for inactivation/removal of HIV, hepatitis A, hepatitis B, and hepatitis C
and also have some effect on parvovirus B19.
Indications for Use
MonoFIX®-VF is indicated for:
■
Treatment and prophylaxis of bleeding associated with factor IX deficiency due
to haemophilia B (Christmas disease)
MonoFIX®-VF is not indicated for the treatment of factor II, VII or X deficiency because
it does not contain therapeutic levels of these coagulation factors. MonoFIX®-VF is
not indicated for the treatment of haemophilia A patients with factor VIII inhibitors.
The number of vials of MonoFIX®-VF to be reconstituted for administration is determined
by dividing the total number of International Units (IU) required by 500 or 1,000
(according to vial size being used), rounded up to the nearest whole number of vials.
Table 5.4: MonoFIX®-VF Dosing Guideline for Haemophilia B
Indication
Dose
Dose
Treatment
Target FVIII
(IU/kg)
Frequency
Duration
(%) (IU/dL)
(per day)
(days)
Prophylaxis1
25-40
2x/week
ongoing
trough 1
Minor haemorrhage
20-30
1
1-2
peak 20-30
Moderate to severe
haemorrhage
■ Haemarthroses
30-50
1-2
1-5
peak 30-50
Life threatening
haemorrhage
■ Intracranial
peak ≥100
haemorrhage
80-100
2
10-12
trough 80-100
Minor surgery2
■ Loading dose
40-60
stat
preoperatively
peak 40-60
■ Maintenance3, 4
15-40
1-2
7-10
trough 20-50
Major surgery
■ Loading dose
70-100
stat
preoperatively
peak 70-100
■ Maintenance3, 4
20-90
1-2
10-12
trough 20-90
1Prophylaxis in children.
2 Includes single tooth extraction. The use of an antifibrinolytic agent such as tranexamic acid is strongly
recommended.
3 Initially (days 1-3) aim for levels at the higher end of this range. Gradually reduce to lower level during
subsequent days.
4An alternative is to use continuous infusion.
Transfusion Medicine Handbook 3rd Edition
Page 61
Precautions
■
Allergic reactions
Allergic reactions or fever are rarely observed. Depending on the nature of an
adverse reaction, the rate of injection should be slowed or stopped to alleviate
symptoms.
■
Antibodies to factor IX Patients with congenital factor IX deficiency may develop neutralising
alloantibodies (inhibitors) to factor IX after treatment. If this occurs specialist
advice must be sought. The reported prevalence for the formation of inhibitors in
patients receiving plasma-derived factor IX is approximately 4%. There has been
no clinical experience with MonoFIX®-VF with respect to inhibitor development
in previously untreated patients.
■
Heparin
MonoFIX®-VF contains 50 - 140 IU heparin in each 500 IU vial and 100 - 280 IU
heparin in each 1000 IU vial. Heparin is known to cause thrombocytopenia and
the possibility of heparin-induced thrombocytopenia (HIT) syndrome should be
considered if thrombocytopenia, with or without thrombosis, develops during
treatment. Consideration should be given to the clinical effect of heparin if high
doses of MonoFIX®-VF are required.
■
Laboratory tests
MonoFIX®-VF is formulated with heparin and antithrombin. Therefore the results
of anticoagulation tests should be interpreted with care.
■
Thrombosis and DIC
High doses of prothrombin complex concentrates (PCC) have been associated
with disseminated intravascular coagulation (DIC). Although MonoFIX®-VF
contains purified factor IX, the potential risk of thrombosis and DIC should be
recognised. The use of products containing factor IX could be hazardous in
patients with a history of fibrinolysis, myocardial infarction, DIC or liver disease.
5.1.3 Prothrombinex®-VF (Factors II, IX and X)
Prothrombinex®-VF is a sterile, freeze-dried powder containing purified human
coagulation factors II, IX, and X. When reconstituted each vial contains 500 IU of
factors IX, approximately 500 IU of factors II and X, and 25 IU antithrombin III, 200 IU
heparin. The product also contains small amounts of factors V and VII.
Prothrombinex®-VF is prepared by adsorption of coagulation factors from plasma onto
an ion-exchange medium followed by selective elution. The manufacturing process
of Prothrombinex®-VF contains dedicated steps including dry heat treatment and
nanofiltration to reduce the potential for viral transmission. The current procedures
are effective for inactivation/removal of HIV, hepatitis A, hepatitis B, and hepatitis C
and may also have some effect on parvovirus B19.
The coagulation factors II, VII, IX and X, synthesised in the liver with the help of vitamin
K, are together commonly called the prothrombin complex.
Isolated congenital deficiency of factor IX is known as haemophilia B. Isolated deficiency
of factor II or factor X is very rare but in severe form can cause a bleeding tendency
similar to that seen in classical haemophilia. Isolated severe deficiency of factor VII
Page 62
Transfusion Medicine Handbook 3rd Edition
leads to reduced thrombin formation and a bleeding tendency due to impaired fibrin
formation and impaired primary haemostasis.
Acquired deficiency of the vitamin K-dependent coagulation factors occurs during
treatment with coumarin vitamin K antagonists such as warfarin. It may also result from
vitamin K deficiency due to malabsorption syndromes, antibiotic therapy, cholestasis
or prolonged parenteral alimentation. If the deficiency progresses, a severe bleeding
tendency results, characterised typically by retroperitoneal or cerebral bleeds rather
than muscle and joint haemorrhage.
Severe hepatic insufficiency also results in markedly reduced levels of the vitamin
K-dependent coagulation factors and may lead to a clinical bleeding tendency. The
haemostatic situation is however often complex due to simultaneous ongoing low-
grade intravascular coagulation, low platelet levels, deficiency of coagulation inhibitors
and disturbed fibrinolysis.
Indications for Use
Prothrombinex®-VF is indicated for:
■
Treatment and perioperative prophylaxis of bleeding associated with acquired
deficiency of prothrombin complex factors such as that caused by treatment
with vitamin K antagonists, or in case of overdose of vitamin K antagonists,
when rapid correction of the deficiency is required
■
Treatment and prophylaxis of bleeding associated with single (or multiple)
congenital deficiency of factors IX, II or X when purified specific coagulation
factor product is not available
Section 6.4:
Oral Anticoagulant – Warfarin Induced Bleeding or Overdose of this
Handbook summarises the updated Australasian Society of Thrombosis and
Haemostasis (ASTH)
Consensus Guidelines for Warfarin Reversal published in the
Medical Journal of Australia, 198 (4): 198-199, 2013. The recommendations for the use
of prothrombin complex concentrate (PCC) in the setting of warfarin-related bleeding
or warfarin overdose have been incorporated in the NZBS app
Reversing Warfarin,
available for iPhone and Android. Consultation with a specialist haematologist or NZBS
Transfusion Medicine Specialist/Medical Officer is recommended.
Table 5.5: Prothrombinex®-VF Dosing Guideline for Haemophilia B
Indication
Dose
Dose
Treatment
Target FIX
(IU/kg)
Frequency
Duration
(%) (IU/dL)
(per day)
(days)
Minor haemorrhage
20-30
1
1-2
peak 20-30
Moderate to severe
haemorrhage
■ Haemarthroses
30-50
1-2
1-5
peak 30-50
Minor surgery
■ Loading dose
40-60
stat
preoperatively
peak 40-60
■ Maintenance
15-40
1-2
7-10
trough 20-50
Transfusion Medicine Handbook 3rd Edition
Page 63
For congenital deficiencies of factors II and X, calculation of the required dose of
Prothrombinex®-VF is based on recovery data obtained for other PCC products
showing that 1 U of factor II or X per kg body weight raises the respective plasma
factor activity by 2% of normal.
Required units (IU) = body weight (kg) x desired rise in factor II or X (as %) x 0.5
Prothrombinex®-VF should not be infused at a rate greater than 3 mL/minute.
Precautions
■
Allergic reactions
Allergic reactions are rarely observed although severe anaphylaxis has been
reported, particularly in patients with factor IX inhibitors. Depending on the
nature of an adverse reaction, the rate of injection should be slowed or stopped
to alleviate symptoms.
■
Antibodies to factor IX
Patients with congenital factor IX deficiency may develop neutralising
alloantibodies (inhibitors) to factor IX after treatment. The reported prevalence
for the formation of inhibitors in patients receiving plasma-derived factor IX is
approximately 4%.
■
Antifibrinolytic agents
Use of Prothrombinex®-VF with epsilonaminocaproic acid or tranexamic acid
is not recommended since only limited data are available on the concomitant
administration of prothrombin complex concentrates and antifibrinolytic agents.
■
Heparin
Prothrombinex®-VF contains 200 IU heparin in each vial. Heparin is
known to cause thrombocytopenia and the possibility of heparin-induced
thrombocytopenia (HIT) syndrome should be considered if thrombocytopenia,
with or without thrombosis, develops during treatment. Consideration should
be given to the clinical effect of heparin if high doses of Prothrombinex®-VF
are required.
■
Thrombosis and DIC
Patients receiving Prothrombinex®-VF, especially at doses greater than 50 IU/
kg of factor IX or following repeated doses, may be predisposed to venous and
arterial thromboembolism, DIC or myocardial infarction. It should be used with
caution in neonates, in who immature hepatic function may lead to delayed
clearance of activated coagulation factors and an increased risk of thrombotic
complications.
5.1.4 FEIBA NF® (Factor VIII inhibitor bypassing fraction)
FEIBA NF® is a sterile, freeze-dried powder of human plasma fraction with factor VIII
inhibitor bypassing activity. FEIBA NF® is available as 20 mL vials containing 500 U
and 1000 U factor VIII inhibitor bypassing activity for reconstitution and intravenous
administration. The potency of FEIBA NF® is expressed in arbitrary units. One Unit
of activity is defined as that amount of FEIBA NF® that shortens the activated partial
thromboplastin time (APTT) of a high titre factor VIII inhibitor reference plasma to 50%
of the blank value.
Page 64
Transfusion Medicine Handbook 3rd Edition
FEIBA NF® is prepared by adsorption of coagulation factors from plasma onto an
ion-exchange medium followed by selective elution. The manufacturing process of
FEIBA NF® contains dedicated steps including nanofiltration and vapour heat treatment
to reduce the potential for viral transmission. The current procedures are effective for
inactivation/removal of HIV, hepatitis B, and hepatitis C and may also have some effect
on hepatitis A and parvovirus B19.
FEIBA NF® is a concentrate of vitamin K-dependent factors in both zymogen and active
form (factors II, IX, and X, mainly non-activated, and factor VII, mainly activated). The
product contains approximately equal unitages of factor VIII inhibitor bypassing activity
and prothrombin complex factors. In addition, 1 - 6 units of factor VIII coagulant antigen
(FVIII C:Ag) per mL are present. The preparation contains only traces of factors of the
kinin generating system. It contains no heparin.
The process of coagulation involves activation of factor X to form Xa, which with
cofactor Va catalyses the formation of thrombin from prothrombin. The production of
sufficient quantities of Xa usually requires a complex of factors VIIIa and IXa. Patients
(often those with haemophilia A or B) can acquire inhibitors to factor VIII or IX during the
treatment with factor VIII or IX replacement therapy, which then prevents the formation
of the complex that catalyses Xa production. FEIBA NF® results in the generation of
Xa and thrombin without the help of the factor VIIIa-IXa complex, thereby bypassing
the inhibitory action of factor VIII (or factor IX) inhibitors.
Indications for Use
FEIBA NF® is indicated for:
■
Routine prophylaxis of bleeding episodes in haemophilia A and B patients
with inhibitors, experiencing ≥ 12 bleeding episodes per year and refractory to
increased dosing with, respectively, factor VIII and IX concentrates
■
Treatment of bleeding episodes and to cover surgical interventions in haemophilia
A and B patients with, respectively, factor VIII and IX inhibitors
■
Longterm use, in combination with factor VIII concentrates, for immune tolerance
induction to eliminate factor VIII inhibitors in patients with haemophilia A, so as
to allow for regular treatment with factor VIII concentrates as in patients without
inhibitors
■
Treatment of severe or life-threatening bleeding episodes in non-haemophiliacs
with acquired inhibitors to factors VIII, XI and XII
Clinical experience suggests that patients with a factor VIII inhibitor titre < 5 Bethesda
units (BU) may be successfully treated with antihemophilic factor (AHF). Patients with
titres ranging between 5 BU and 10 BU may either be treated with AHF or FEIBA
NF®. Patients with factor VIII inhibitor titres > 10 BU have generally been refractory to
treatment with AHF. However, since a single dose of FEIBA NF® contains considerably
less factor VIII than factor VIII concentrate, FEIBA NF® is considered the treatment of
choice in high responder patients, even if the current inhibitor titre is low.
Transfusion Medicine Handbook 3rd Edition
Page 65
Table 5.6: Guideline for Treatment of Patients with Haemophilia A and Factor VIII
Inhibitors
Inhibitor Titre
Response to
Minor /
Severe / Life-
(BU1
/mL)
FVIII
Moderate
threatening Bleeding
Bleeding
or Surgery
< 5
low responder
FVIII or FEIBA NF®
FVIII or FEIBA NF®
> 5
high responder
FEIBA NF®
FEIBA NF®
5 - 10
low responder
FVIII or FEIBA NF®
FEIBA NF®
5 - 10
high responder
FEIBA NF®
FEIBA NF®
< 10
low responder
FEIBA NF®
FEIBA NF®
> 10
high responder
FEIBA NF®
FEIBA NF®
1 Bethesda Unit is defined as that amount of antibody that will inhibit 50% of the factor VIII activity of fresh
plasma after incubation for 2 hours at 37 °C.
Dosage and Administration
Dosage is independent of the patient’s inhibitor titre. As a general rule, a dose of 50
- 100 U FEIBA NF® per kilogram body weight is recommended. However, due to the
risk of thrombosis, the total daily dose should not exceed 200 U/kg and, for any one
dose, the infusion rate should not exceed 2 U/kg per minute. Due to varying response
to treatment the following dosage recommendations are only guidelines.
Table 5.7: FEIBA NF® Dosing Guideline
Bleeding
Dose1
Dose Frequency Treatment Duration
Indication
(U/kg)
(day)
Prophylaxis2
70-100
alternate day
ongoing
Surgery
50-100
4
preoperatively and until
wound healing
Mucous membrane3
50
4
until clinical improvement
Joint, muscle, soft
50-75
2
until clinical improvement
tissue - mild to
moderate
Joint, muscle, soft
100
2
until clinical improvement
tissue - severe
Other severe4
100
2
until clinical improvement
e.g., CNS
1Single doses of 100 U/kg and daily doses of 200 U/kg body weight should not be exceeded.
2Dose recommendations, based on body weight, are the same for paediatric patients as for adults.
3 If bleeding is not controlled the dose may be increased to 100 U/kg, provided the maximum daily dose
is not exceeded.
4 In individual cases the frequency may be increased to 6-hourly until clinical improvement, provided the
maximum daily dose is not exceeded.
Page 66
Transfusion Medicine Handbook 3rd Edition
Contraindications
FEIBA NF® is contraindicated in cardiac surgery involving cardiopulmonary bypass
and procedures involving extracorporeal membrane oxygenation (ECMO) due to the
high risk of thrombotic adverse events.
Precautions
■
Allergic reactions
FEIBA NF® has been associated with severe allergic and anaphylactoid reactions.
■
Anamnestic response
Administration of FEIBA NF® to patients with inhibitors may result in an initial
anamnestic rise in inhibitor levels. Clinical data suggest that the efficacy of FEIBA
NF® is not reduced. Inhibitor levels may decrease over time with continued
administration of FEIBA NF®.
■
Laboratory tests
In vitro tests such as APTT, whole blood clotting time (WBCT) and
thromboelastography (TEG) used to monitor efficacy may not correlate with
clinical improvement. Attempts to normalise these values by increasing the
dose of FEIBA NF® are discouraged and may induce DIC through overdosage.
■
Passive anti-HBs transfer
Administration of FEIBA NF® with a transitory rise of passively transferred
hepatitis B surface antibodies may mislead the interpretation of positive serology
test results.
■
Sodium
The maximum daily dose of FEIBA NF® may contain > 200 mg sodium and this
should be taken into consideration for patients on a sodium-restricted diet and/
or with renal impairment.
■
Thrombosis
Patients should not receive single doses of FEIBA NF® > 100 U/kg body weight
or daily doses > 200 U/kg body weight as these may predispose venous and
arterial thromboembolism, DIC, myocardial infarction or stroke. Patients receiving
doses such as these should be monitored for the development of adverse events.
The possible presence of risk factors for thromboembolism, even in patients
with haemophilia, should always be considered.
Interactions with Other Medicines
The use of tranexamic acid, an antifibrinolytic agent, in combination with FEIBA NF® is
not recommended due to an increased risk of thrombotic events. If treatment with both
is indicated the products should be administered at least 12 hours apart. Concomitant
use with recombinant factor VIIa may potentially result in an adverse thrombotic event.
5.1.5 RiaSTAP® (Fibrinogen)
RiaSTAP® does not have full New Zealand registration and is in limited supply so
consultation with an NZBS Transfusion Medicine Specialist/Medical Officer is required
prior to release of this product.
RiaSTAP® is a freeze-dried powder of purified human fibrinogen (factor I). Each 50 mL
vial when reconstituted contains 1 g of human fibrinogen for intravenous administration.
Transfusion Medicine Handbook 3rd Edition
Page 67
Indications for Use
RiaSTAP® is indicated for:
■
Prophylaxis and treatment of bleeding in patients with congenital fibrinogen
deficiency including afibrinogenaemia and hypofibrinogenaemia
Dosage and Administration
The (functional) fibrinogen level should be determined in order to calculate individual
dosage requirements. The frequency of administration should be determined on an
individual patient basis by regular measurement of plasma fibrinogen and continuous
monitoring of the clinical condition of the patient. Dosage recommendations in the
treatment of children are the same as for adults.
Dose of RiaSTAP® (mg/kg body weight) = target level (g/L) – measured level (g/L)
0.017 (g/L per mg/kg body weight)
If the serum fibrinogen level is not known the recommended dose is 70 mg per kilogram
body weight administered intravenously.
As a guide for subsequent dosing the target level of 1 g/L for minor events should
be maintained for three days. The target level of 1.5 g/L for major events should be
maintained for seven days.
Precautions
■
Thrombosis
There is a risk of thrombosis when patients with congenital fibrinogen deficiency
are treated with human fibrinogen, particularly with high or repeat doses.
Patients, particularly those with risk factors for thromboembolic disease including
neonates, should be monitored closely for signs and symptoms of thrombosis.
■
Serum sodium
RiaSTAP® contains up to 164 mg (7.1 mmol) sodium per vial and this should
be taken into consideration for patients on a sodium-restricted diet and/or with
renal impairment.
Adverse Events
The following adverse reactions have been reported from clinical studies.
Table 5.8: Adverse Events Associated with Administration of RiaSTAP®
Event
Frequency
Urticaria, rash, fall in blood pressure, dyspnoea
≥ 1/10,000 and < 1/1,000
Increase in body temperature
≥ 1/10,000 and < 2/1,000
Headache
≥ 1/10,000 and < 1/1,000
Thromboembolic episodes
< 1/10,000
5.1.6 Fibrogammin® P (Factor XIII)
Fibrogammin® P does not have full New Zealand registration and so consultation with an
NZBS Transfusion Medicine Specialist/Medical Officer is required prior to product release.
Page 68
Transfusion Medicine Handbook 3rd Edition
Fibrogammin® P is a freeze-dried powder of purified human factor XIII. Each vial when
reconstituted contains 250 units of factor XIII.
Indications for Use
Fibrogammin® P is indicated for:
■
The prophylaxis and treatment of bleeding associated with congenital FXIII
deficiency
■
Supportive therapy in the case of disturbance in wound healing associated with
congenital FXIII deficiency, especially with venous ulcers or following extensive
surgery
■
The treatment of bleeding associated with acquired FXIII deficiency
Dosage and Administration
The following guideline is to assist in the immediate management of a patient until
consultation with a specialist haematologist or NZBS Transfusion Medicine Specialist/
Medical Officer. The exact loading and maintenance dose and dosing interval should be
based on the patient’s clinical condition and response to therapy. It is recommended to
monitor the increase in factor XIII activity. For major surgery and severe haemorrhage
the aim is to obtain normal values.
Table 5.9: Fibrogammin® P Dosing Guideline
Indication
Dose
Treatment Duration
(IU/kg)
Congenital FXIII
deficiency
Prophylaxis
10
approx. once per month,
individualised to bleeding frequency
Moderate to severe
10-20
daily for severe haemorrhage and
haemorrhage1
until bleeding stops
Surgery
■ Loading dose
35
preoperatively
■ Maintenance
10
daily until complete wound healing
Acquired FXIII
deficiency
Moderate to severe
15-20
daily until bleeding stops or normal
haemorrhage
FXIII levels achieved spontaneously
Disorder of wound
healing
Surgery2
10
preoperatively and then once daily
for 3 days
1 Severe life threatening bleeding may initially require doses up to 50 IU/kg with an aim to achieve normal
factor XIII levels.
2In high-risk patients the dose can be increased to 15 - 20 IU/kg.
Transfusion Medicine Handbook 3rd Edition
Page 69
Precautions
■
Allergic reactions
Allergic reactions or fever are rarely observed. Depending on the nature of an
adverse reaction, the rate of injection should be slowed or stopped to alleviate
symptoms.
■
Antibodies to factor XIII Patients with congenital deficiency may rarely develop neutralising alloantibodies
(inhibitors) to factor XIII after repeated treatment.
■
Thrombosis In cases of fresh thrombosis, caution should be exercised due to the fibrin-
stabilising effect.
5.2
Natural Inhibitors of Coagulation
5.2.1 Thrombotrol®-VF (Antithrombin III)
Thrombotrol®-VF is a sterile, freeze-dried powder of purified human antithrombin
III (ATIII). Each vial when reconstituted contains 1000 IU of ATIII for intravenous
administration.
Indications for Use Thrombotrol®-VF is indicated in patients with hereditary deficiency of antithrombin
under the following circumstances:
■
Prophylactic administration for the prevention of thromboembolism in surgery,
pregnancy and during childbirth
■
Therapeutic administration in thrombosis or pulmonary embolism
The dose should be based on pretreatment and desired ATIII levels. The dose can
be calculated using the following formula which is based on an incremental in-vivo
recovery of ATIII of 2.2% per IU/kg bodyweight using a functional ATIII assay.
Dose (IU) = [Desired ATIII (IU) - Pretreatment ATIII level*(IU)] × Wt (kg)
2.2
* expressed as % normal level based on functional ATIII assay
Under conditions of acute consumption, the biological half-life of ATIII may be reduced
from 2.8 days to only a few hours. Fol owing acute thrombosis where ATI I levels should
be maintained ≥ 100% for 2 - 8 days, plasma ATIII levels determined several times per
day may be used to guide replacement therapy.
ATIII replacement may also be given prophylactically to overcome heparin resistance
and reduce the risk for circuit thrombosis in patients (particularly neonates) undergoing
extracorporeal membrane oxygenation (ECMO) during cardiac surgery. In this
situation acute consumption of ATIII may lead to low plasma ATIII levels and failure to
achieve adequate anticoagulation despite increasing heparin infusion doses. ATIII is
administered as either intermittent intravenous bolus doses, each over 10 - 20 minutes,
or as a continuous infusion with the aim to maintain an activated clotting time (ACT) >
400 seconds together with a target ATIII level of 60 - 100%. The doses required are
typically larger than those used for replacement in hereditary deficiency.
5.2.2 Ceprotin (Protein C)
Ceprotin does not have full New Zealand registration and so consultation with an NZBS
Transfusion Medicine Specialist/Medical Officer is required prior to product release.
Page 70
Transfusion Medicine Handbook 3rd Edition
Ceprotin is a sterile, freeze-dried powder of purified human protein C. Each vial when
reconstituted contains 1000 IU of protein C for intravenous administration.
Indications for Use
Ceprotin is indicated for:
■
Prophylaxis and treatment of venous thrombosis and purpura fulminans in
paediatric and adult patients with severe congenital protein C deficiency
Dosage and Administration
The loading and maintenance dose and dosing interval should be based on the clinical
condition, the severity of the protein C deficiency (i.e., pretreatment level) and desired
protein C level. It is recommended to monitor the increase in protein C activity. These
guidelines are recommended for neonatal, paediatric and adult patients.
Table 5.10: Ceprotin Dosing Guideline1
Indication
Initial Subsequent Maintenance Target Protein C
Dose
Doses
Dose
(%) (IU/dL)
(IU/kg)
(IU/kg)
(IU/kg)
Acute episode/
100-120
60-80
45-60
peak 100
Short term
6 hourly for
6-12 hourly
prophylaxis2
three doses
Long term
-
-
45-60
trough >25
prophylaxis
12 hourly
1Dosing should be adjusted according to the pharmokinetic profile for each individual.
2Ceprotin should be continued until desired anticoagulation is achieved.
Adverse Reactions
Common reactions include rash, itch and lightheadedness. Other reported reactions
include restlessness and hyperhydrosis.
Precautions
■
Allergic reactions
Ceprotin may contain traces of mouse protein and/or heparin, to which allergic
reactions cannot be ruled out.
■
Heparin
Ceprotin contains trace amounts of heparin which may lead to heparin-induced
thrombocytopenia (HIT).
■
Sodium
Ceprotin contains > 200 mg sodium and this should be taken into account for
patients on a sodium-restricted diet and/or with renal impairment.
5.3
Albumin Solutions
5.3.1 Albumex® 4 (Human albumin 4%)
Albumex® 4 contains 40 g/L albumin in solution for intravenous injection and is iso-
oncotic with human serum. It is available in vials of 50 mL and 500 mL volume. When
Transfusion Medicine Handbook 3rd Edition
Page 71
infused into adequately hydrated patients its effect is to expand the circulating blood
volume by an amount approximately equal to the volume of Albumex® 4 infused. It is
prepared by a combination of Cohn cold-ethanol fractionation and chromatography.
The manufacturing process of Albumex® 4 includes pasteurisation and cold
temperature incubation to reduce the potential for viral transmission. The current
procedures are effective for inactivation/removal of HIV, hepatitis A, hepatitis B, and
hepatitis C and may also be of limited value against parvovirus B19.
Indications for Use
Table 5.11: Clinical Indications for Use of Albumex® 4
Indication
Comments
Hypovolaemia / Shock
■
Preservation of an adequate circulating blood volume
should be the primary aim of therapy. Albumex® 4
may be the initial plasma expander of choice for
shock associated with significant hypoalbuminaemia
(plasma albumin < 25 g/L).
■
May also be useful following initial resuscitation with
crystalloid or synthetic colloid solutions in patients in
whom extended support of the intravascular volume
is required such as seriously ill patients with multiple
organ failure or the systemic capillary leak syndrome.
■
The use of albumin for fluid resuscitation of patients
with traumatic brain injury is not recommended.
Cardiopulmonary
■
Priming the pump for cardiopulmonary bypass
Bypass
surgery for patients with poor left ventricular function
and other complicating factors such as long bypass
time, anaemia or repeat surgery.
Plasma Exchange
■
Indicated as a replacement solution in plasma
exchange procedures, particularly when the volume
exchanged exceeds 20 mL/kg body weight.
■
In patients with thrombotic thrombocytopenic
purpura, fresh frozen plasma or cryosupernatant may
be the preferred replacement solution.
Precautions
■
Adverse effects
Adverse reactions to albumin solutions are uncommon and are usually mild and
transient. Chills, fever, urticaria, flushing, nausea, headache and dyspnoea may
occur. More serious allergic events including hypotension and anaphylaxis are
reported. In addition, hypotension has been reported in patients given albumin
who are on angiotensin-converting enzyme (ACE) inhibitors.
■
Aluminium accumulation
Albumex® 4 contains trace amounts of aluminium (200 µg/L). Accumulation
of aluminium in patients with chronic renal insufficiency has led to toxic
manifestations such as hypercalcaemia, vitamin D-refractory osteodystrophy,
anaemia and severe progressive encephalopathy. Therefore, when large
Page 72
Transfusion Medicine Handbook 3rd Edition
volumes of albumin are contemplated for administration to such patients,
serious consideration should be given to these potential risks relative to the
anticipated benefits.
■
Circulatory overload
Circulatory overload can be avoided by monitoring the rate and volume of
infusion. Patients with cardiac failure, renal insufficiency, stabilised chronic
anaemia or on cardiopulmonary bypass are at special risk of developing
circulatory overload.
■
Compatibility with other fluids
Albumex® 4 should not be mixed with protein hydrolysates, amino acid solutions,
solutions containing alcohol, or solutions containing drugs that bind to albumin
such as calcium channel blockers.
■
Shock Administration of albumin can aggravate myocardial depression when present
in patients with shock.
Safety of Albumin for Fluid Resuscitation
In 2011 the Cochrane Injuries Group reported results of a meta-analysis of the available
medical literature on “Human albumin solution for resuscitation and volume expansion
in critically ill patients” and concluded that for patients with hypovolaemia there is no
evidence that albumin reduces mortality when compared with cheaper alternatives
such as saline. Contributing significantly to this report were results from the SAFE
trial (Saline vs Albumin Fluid Evaluation) which compared the safety and efficacy of
albumin versus saline in Australasian intensive care units. The study concluded that
albumin and saline should generally be considered clinically equivalent treatments for
intravascular resuscitation in the ICU although further study is required for more highly
selected populations of critically ill patients, for example those with brain injury. The
Cochrane report also concluded that for patients with burns and hypoalbuminaemia
there is no evidence that albumin reduces mortality. Due to the increased cost of
albumin compared to alternatives such as saline, albumin should only be used within
the context of further trials.
5.3.2 Albumex® 20 (Human albumin 20%)
Albumex® 20 contains 200 g/L albumin in solution for intravenous injection and is
hyperoncotic and hypo-osmotic compared to human serum. It is available in vials
of 10 mL and 100 mL volume. When infused it supplies the oncotic equivalent
of approximately four times its volume of human plasma. Albumex® 20 has two
main functions: maintenance of plasma colloid osmotic pressure and transport of
intermediate products in the transport and exchange of tissue metabolites. It is
prepared by a combination of Cohn cold-ethanol fractionation and chromatography.
The manufacturing process of Albumex® 20 includes pasteurisation and cold
temperature incubation to reduce the potential for viral transmission. The current
procedures are effective for inactivation/removal of HIV, hepatitis A, hepatitis B, and
hepatitis C and may also be of limited value against parvovirus B19.
Transfusion Medicine Handbook 3rd Edition
Page 73
Indications for Use
Table 5.12: Clinical Indications for Use of Albumex® 20
Indication
Comments
Hypoproteinaemia in ■ Administered when there are (or it is anticipated) clinical
acutely ill patients
problems or complications from reduced oncotic
pressure, and/or as an adjunct to diuretic therapy.
Shock
■
May be used for the resuscitation of patients in shock
due to acute loss of blood or plasma however 4%
human albumin is preferred when available.
Burns
■
Extensive burns are followed by sequential shifts in the
distribution of body water, salt and proteins resulting in
hypovolaemic shock and circulatory failure.
■
Initially there is an increased vascular permeability
leading to loss of water and proteins into
the extravascular compartment leading to
haemoconcentration.
■
Large volumes of crystalloid solutions should be infused
to restore the constricted intravascular fluid space
while smaller amounts of Albumex® 20 are required to
maintain adequate plasma volume and colloid osmotic
pressure.
Haemodialysis
■
May be used to assist with the rapid removal of excess
extravascular fluid and to maintain perfusion pressure.
Adult Respiratory
■
ARDS, an acute inflammatory lung injury with diffuse
Distress Syndrome
alveolar damage and increased vascular permeability, is
characterised by inadequate oxygenation secondary to
non-cardiogenic pulmonary oedema.
■
Combination therapy with Albumex® 20 and diuretics
may improve fluid balance, oxygenation, and
haemodynamics.
■
In patients who have undergone abdominal surgery,
intravenous administration of 20% albumin immediately
after the operation has been shown to improve lung
compliance and gaseous exchange.
Therapeutic plasma
■
TPE is a procedure in which approximately one plasma
exchange
volume is exchanged with a colloid replacement
solution. The choice of replacement fluid and its
concentration are determined by the particular clinical
situation and the frequency of the procedure.
■
Iso-oncotic 4% albumin solution is the preferred
replacement material. If the patient’s serum albumin
level is not maintained 20% albumin may be indicated. If
exchange occurs less frequently than once a week, less
concentrated colloids may be appropriate.
Page 74
Transfusion Medicine Handbook 3rd Edition
Precautions
■
Adverse effects
Adverse reactions to albumin solutions are uncommon and are usually mild and
transient. Chills, fever, urticaria, flushing, nausea, headache and dyspnoea may
occur. More serious allergic events including hypotension and anaphylaxis are
reported. Hypotension has been reported in patients given albumin who are on
angiotensin-converting enzyme (ACE) inhibitors.
■
Aluminium accumulation
Albumex® 20 contains trace amounts of aluminium (≤ 200 µg/L). Accumulation
of aluminium in patients with chronic renal insufficiency has led to toxic
manifestations such as hypercalcaemia, vitamin D-refractory osteodystrophy,
anaemia and severe progressive encephalopathy. Therefore, when large
volumes of albumin are contemplated for administration to such patients,
serious consideration should be given to these potential risks relative to the
anticipated benefits.
■
Circulatory overload
The colloid osmotic effect of Albumex® 20 is approximately four times that of
plasma. Circulatory overload can be avoided by monitoring the rate and volume
of infusion. Patients with cardiac failure, renal insufficiency or stabilised chronic
anaemia often have an increased circulatory plasma volume and are at special
risk of developing circulatory overload.
■
Compatibility with other fluids
Albumex® 20 should not be mixed with protein hydrolysates, amino acid
solutions, solutions containing alcohol, or solutions containing drugs that bind
to albumin such as calcium channel blockers.
■
Hyperoncotic effects
As Albumex® 20 is hyperoncotic, it must be diluted with or followed by crystalloid
solution in the presence of dehydration or shock.
If Albumex® 20 is diluted to an iso-oncotic protein concentration (4-5% albumin)
prior to administration, this must be done with an iso-osmotic electrolyte solution
such as 0.9% saline. Under no circumstances should water be used since the
lower tonicity will lead to intravascular haemolysis.
■
Hypoproteinaemia
The infusion of Albumex® 20 is not justified in hypoproteinaemic states associated
with chronic cirrhosis, malabsorption, protein losing enteropathies, pancreatic
insufficiency, undernutrition. Albumex® 20 may be indicated as a temporising
measure in selected cases awaiting liver transplantation.
■
Nephrosis
In chronic nephrosis, infused albumin is promptly excreted by the kidneys with
no or limited relief of the chronic oedema. Albumex® 20 may be indicated as a
temporising measure in selected cases awaiting renal transplantation.
■
Shock Administration of albumin can aggravate myocardial depression when present
in patients with shock.
Transfusion Medicine Handbook 3rd Edition
Page 75
■
Sodium levels The sodium levels in this product are 48 - 100 mmol/L. This should be noted
when the product is used in patients requiring sodium restriction.
5.4
Immunoglobulin Preparations
General Considerations
Immunoglobulin products are sterile, preservative-free solutions of concentrated IgG
prepared by Cohn cold-ethanol fractionation and chromatography of human plasma.
In the case of specific immunoglobulins, for example anti-D immunoglobulin and
hepatitis B immunoglobulin, plasma is obtained from individuals with a high titre of
the required antibody.
The manufacturing process contains two or more viral removal steps such as
pasteurisation (inactivation) and viral filtration (nanofiltration) reducing the possibility of
virus transmission. Currently available immunoglobulins have not been implicated in the
transmission of viral infectious diseases including human immunodeficiency virus (HIV).
Precautions
■
Immunoglobulin products approved for intramuscular injection must not be
administered intravenously because of the potential for severe adverse reactions.
They should be given slowly by deep intramuscular injection using an appropriate
sized needle and care should be taken to draw back on the plunger of the syringe
before injection in order to be certain that the needle is not in a blood vessel.
■
If an intramuscular dose of more than 5 mL is required, it is advisable to
administer in divided doses at different sites. Hyaluronidase and/or a suitable
local anaesthetic may be added to the injection if desired. When large doses
of immunoglobulins are required, consider using an alternative product suitable
for intravenous administration.
■
Intramuscular injection of immunoglobulin products should be avoided in
patients with a low platelet count. In these circumstances they may be given
subcutaneously.
■
Immunoglobulin products approved for subcutaneous infusion must not be
administered intravenously because of the potential for severe adverse reactions.
■
Immunoglobulin products should be given with caution to patients with a
history of prior systemic allergic reactions following the administration of human
immunoglobulin preparations. In the case of allergic shock, treatment should
follow recommended guidelines for managing anaphylaxis.
Contraindications
Immunoglobulin products are contraindicated in individuals:
■
With isolated immunoglobulin A (IgA) deficiency, unless they have been tested
and shown not to have circulating anti-IgA antibodies.
■
Who have severe thrombocytopenia or any coagulation disorder that would
contraindicate intramuscular injection.
Page 76
Transfusion Medicine Handbook 3rd Edition
Interactions with Other Drugs
■
Immunoglobulin should not be mixed with other pharmaceutical products,
except as indicated by the manufacturer.
■
Passively acquired antibody can interfere with the response to live, attenuated
virus vaccines. Therefore, administration of such vaccines, e.g., measles and
varicella, should be deferred for at least 3 months after passive immunisation
with immunoglobulin preparations and antibody-containing blood components,
i.e., whole blood, resuspended red cells, plasma and platelets.
General
Recommendations on Immunization from the Advisory Committee on
Immunization Practices (ACIP), including recommended deferral intervals, are
available from the Centers for Disease Control and Prevention (CDC) at www.
cdc.gov/vaccines/. Consultation with a NZBS Transfusion Medicine Specialist/
Medical Officer is recommended.
■
By the same token, immunoglobulins and antibody-containing blood components
should not be administered for at least two weeks after a live vaccine has been
given.
■
Inactivated vaccines may be administered concurrently with passive antibody
(although in separate syringes) to induce active immunity, as is sometimes done
for tetanus-prone wounds.
Passive Transfer of Antibodies and Interference with Serological Testing
After injection of immunoglobulin, the transient rise of the various passively transferred
antibodies in the patient’s blood may result in misleading positive results in serological
testing. Although it has not been determined whether or not immunoglobulin products
can transmit human parvovirus B19, they are known to contain antibodies to the virus.
Adverse Reactions
■
Local tenderness, erythema and stiffness may occur at the site of injection and
may persist for several hours. This may occur after any intramuscular injection.
■
Mild pyrexia, malaise, drowsiness and urticaria have occasionally been reported
after injections of immunoglobulins.
■
True allergic responses are rare. Skin lesions, headache, dizziness, nausea,
generalised hypersensitivity reactions and convulsions have been reported on
rare occasions. These occur more frequently in patients who have a profound
hypogammaglobulinaemia.
5.4.1 Normal Immunoglobulin-VF
Normal Immunoglobulin-VF is a sterile, preservative-free solution containing 160 mg/
mL human plasma proteins with at least 98% being immunoglobulin (mainly IgG).
Normal Immunoglobulin-VF is intended for intramuscular injection.
Transfusion Medicine Handbook 3rd Edition
Page 77
Indications for Use
Table 5.13: Clinical Indications for Use of Normal Immunoglobulin-VF
Indication
Comments
Primary
Congenital and acquired forms.
hypogammaglobulinaemia
Secondary
Acquired hypogammaglobulinaemia, as seen in
hypogammaglobulinaemia haematological malignancies, nephrosis or protein-
losing enteropathy, complicated by recurrent
infections.
Hepatitis A
Routine passive protection is recommended in those
exposed less than two weeks previously for the
following categories of individuals:
■
Household contacts of an index case who have
not already had hepatitis A or have no evidence of
immunity to the virus
■
Common source exposures (i.e., a vehicle such
as food or water is identified as a common source
of infection for multiple cases of hepatitis) where
administration of Normal Immunoglobulin-VF
should be considered for all those exposed
■
Institutional contacts
■
Staff in institutions where hepatitis is endemic
■
Newborn infants where the mother has developed
acute hepatitis A from two weeks before birth to
one week after birth
Routine prophylaxis is not recommended for school,
factory or hospital contacts. Active immunisation
with hepatitis A vaccine is recommended in these
circumstances.
Vaccination is also recommended for those at risk of
exposure to hepatitis A, such as persons travelling to
areas of high or intermediate endemicity and staff at
child day care centres.
Immunoglobulin is no longer routinely recommended
for pre-travel use however may be appropriate for
providing optimal protection to adults aged over 40
years of age, immunocompromised individuals and
those with chronic medical conditions including liver
disease who are planning to depart in < 2 weeks.
Travellers under 12 months of age and those unable
or unwilling to receive vaccination should also receive
immunoglobulin.
Page 78
Transfusion Medicine Handbook 3rd Edition
Table 5.13: Clinical Indications for Use of Normal Immunoglobulin-VF continued
Indication
Comments
Rubella
Although Normal Immunoglobulin-VF can prevent
or modify the clinical disease in susceptible rubella
contacts if given within 72 hours of exposure, it does
not prevent viraemia in such patients. It should not be
relied upon to prevent congenital malformations due to
rubella if given to susceptible pregnant women during
the first trimester. Therefore, routine use of Normal
Immunoglobulin-VF for post-exposure prophylaxis of
rubella in early pregnancy is not recommended. It may
be considered if termination of the pregnancy is not
an option but termination must be discussed when
maternal infection is confirmed.
Measles
Recommended for the following contacts of measles
cases:
■
Immune-compromised or immune-deficient
children
■
Pregnant women
■
Immune-competent children aged under 15
months beyond 72 hours after exposure
■
People outside the 72-hour window for MMR who
have not had a history of measles infection or
vaccination
For these individuals Normal Immunoglobulin-VF is
given to attenuate disease and should be given as
soon as possible, to a maximum of six days after
exposure.
Poliomyelitis
Normal Immunoglobulin-VF is recommended for
susceptible contacts not previously immunised against
poliomyelitis.
In general, the earlier in the incubation period of hepatitis A, rubella, measles or
poliomyelitis that normal immunoglobulin is given, the greater its effectiveness.
Transfusion Medicine Handbook 3rd Edition
Page 79
Dosage and Administration
Table 5.14: Dosage Recommendations for Normal Immunoglobulin-VF
Indication
Dose
Dose Interval (months)
(mL/kg)
Hepatitis A
■
Short-term prophylaxis (general)
0.03
■
Long-term prophylaxis (general)
0.06
5 monthly1
■
Short term travel (< 3 months)
0.03
■
Long term travel2 (> 3 months)
0.06
Repeat 4-6 monthly
■
Institutional contacts2
0.06
■
Institutions where hepatitis A endemic2
0.06
Repeat 6 monthly
Hypogammaglobulinaemia
0.6
Additional loading dose in
first month, then monthly
Measles prophylaxis3
0.6
Poliomyelitis
0.3
Rubella
0.2
1Perform serological checks to assess if active immunity has developed.
2 Use of hepatitis A vaccine may be more appropriate for these individuals provided there is sufficient time
for active immunity to develop (7–10 days).
3 0.6 mL/kg to a maximum dose of 5 mL in immune-competent infants aged under 15 months and to a
maximum dose of 15 mL, recommended as three 5 mL injections, in pregnant women, immune-competent
adults and immune-compromised or immune-deficient children.
5.4.2 Hepatitis B Immunoglobulin-VF
Hepatitis B Immunoglobulin-VF is a sterile, preservative-free solution containing not less
than 100 IU/mL neutralising hepatitis B antibodies. Donations used in the preparation
of Hepatitis B Immunoglobulin-VF are selected on the basis that they contain high
levels of specific antibodies against HBsAg. Hepatitis B Immunoglobulin-VF is provided
as 400 IU vials, intended for intramuscular injection.
Indications for Use
Hepatitis B Immunoglobulin-VF is indicated for:
■
Post-exposure prophylaxis in persons who did not receive prior vaccination or
whose vaccination regimen is incomplete, or when the hepatitis B antibody level
is inadequate (< 10 IU/L)
■
Infants born to HBsAg-positive mothers, either chronic carriers or those who
contract hepatitis B during pregnancy
■
Patients with hepatitis B undergoing a liver transplant, to protect the transplanted
liver
Post-exposure prophylaxis should be considered fol owing percutaneous or
permucosal exposure to HBsAg-positive or suspected HBsAg-positive material, for
example, following needle stick injury, oral ingestion or sexual exposure.
Page 80
Transfusion Medicine Handbook 3rd Edition
Dosage and Administration
For maximum protective effect, Hepatitis B Immunoglobulin-VF should be given within
72 hours of exposure. Efficacy is greatly reduced if it is given after a longer interval.
Table 5.15: Prophylaxis with Hepatitis B Immunoglobulin-VF in Adults Following
Percutaneous or Permucosal Exposure to HBsAg-positive or Suspected
HBsAg-positive Material
Source material
Vaccination history
No prior vaccination or
Completed vaccination
incomplete vaccination
regimen
regimen
Confirmed
A single dose of 400 IU
Test for anti-HBs. If level <10
positive for
hepatitis B immunoglobulin
IU/L, give a single dose of 400
HBsAg
immediately and initiate
IU hepatitis B immunoglobulin
hepatitis B vaccination
immediately plus a booster
regimen at the same time.
vaccination.
High risk for
Initiate hepatitis B vaccination
Test exposed person for HBs
HBsAg but not
regimen. Test source material
antibody. If level <10 IU/L, test
confirmed
for HBsAg and, if positive,
source material for HBsAg
give a single dose of 400 IU
and, if positive, give a single
hepatitis B immunoglobulin.
dose of 400 IU Hepatitis B
Immunoglobulin-VF plus a
booster vaccination.
Uncertain or low
Initiate hepatitis B vaccination
Nothing required.
risk
regimen.
Active immunisation with hepatitis B vaccine should always be commenced in
conjunction with administration of Hepatitis B Immunoglobulin-VF in patients exposed
to hepatitis B virus. Vaccination should be initiated simultaneously with the passive
immunoglobulin, but administered at a different site.
Prophylaxis in Infants Born to HBsAg-positive Mothers
Where HyperHEP™ S/D is unavailable, give 100 IU Hepatitis B Immunoglobulin-VF
to the infant at birth and initiate simultaneously a hepatitis B vaccination regime
administered at a different site.
5.4.3 HyperHEP™ S/D
HyperHEP™ S/D is a sterile, preservative-free, solvent/detergent-treated solution
containing not less than 220 IU/mL neutralising hepatitis B antibodies. Donations used
in the preparation of HyperHEP™ S/D are selected on the basis that they contain high
levels of specific antibodies against HBsAg. HyperHEP™ S/D is supplied as a 0.5 mL
neonatal single-dose syringe containing at least 110 IU of hepatitis B immunoglobulin,
intended for intramuscular injection.
Indications for Use
HyperHEP™ S/D is indicated for:
■
Infants born to HBsAg-positive mothers, either chronic carriers or those who
contract hepatitis B during pregnancy
Transfusion Medicine Handbook 3rd Edition
Page 81
Dosage and Administration
The infant should receive a single 110 IU neonatal dose of HyperHEP™ S/D at birth.
The dose is preferably given within 12 hours of birth as efficacy decreases markedly
if treatment is delayed beyond 48 hours. Hepatitis B vaccine should be administered
concurrently with hepatitis B immunoglobulin (or at most within 7 days) but at a
separate site. If administration of the first dose of hepatitis B vaccine is delayed for
as long as 3 months, then a 0.5 mL dose of HyperHEP™ S/D should be repeated
at 3 months. If vaccine is refused, the 0.5 mL dose of HyperHEP™ S/D should be
repeated at 3 and 6 months.
5.4.4 Tetanus Immunoglobulin-VF
Tetanus Immunoglobulin-VF is a sterile, preservative-free, pasteurised solution with a
tetanus antitoxin activity of not less than 100 IU/mL. Donations used in the preparation
of tetanus immunoglobulin are selected on the basis that they contain high levels of
specific antibodies against the toxin of Clostridium tetani. Tetanus Immunoglobulin-VF
is intended for intramuscular injection.
Indications for Use
Tetanus Immunoglobulin-VF is indicated for:
■
Passive protection of individuals who have sustained a tetanus-prone wound
and who have either not been actively immunised against tetanus or whose
immunisation history is doubtful
■
Passive protection of fully immunised individuals with a tetanus-prone wound if
more than 10 years have elapsed since the last dose of tetanus toxoid vaccine
In both of the above instances, active immunisation with tetanus vaccine should be
commenced at the same time. Although tetanus immunoglobulin and vaccine can
be given at the same time, they should be administered in opposite limbs, using
separate syringes.
Page 82
Transfusion Medicine Handbook 3rd Edition
Table 5.16: Guide to Tetanus Prophylaxis in Wound Management
Type of wound
History
Clean, minor wound
All other wounds
of active
immunisation
Tetanus
Tetanus
Tetanus
Tetanus
Vaccine
Immunoglobulin-
Immunoglobulin-
1
VF
Vaccine1
VF
Never
immunised or
Yes
No
Yes
Yes
< 3 doses
Immunised and
≥ 3 doses:
< 5 years since
No
No
No
No
last dose
5-10 years
No
No
Yes
No
since last dose
> 10 years
Yes
No
Yes
Yes
since last dose
1 Children <
8 years old receive combined DTPa vaccine. Persons 8 years and older receive combined
dT vaccine.
Dosage and Administration
Good medical care is essential in the prevention of tetanus from fresh wounds.
Thorough cleansing and removal of all foreign and necrotic material from the site of
injury is important.
The minimum routine prophylactic dose of Tetanus Immunoglobulin-VF for adults or
children is 250 IU given slowly by deep intramuscular injection. The dose should be
doubled to 500 IU if the wound is grossly contaminated or if more than 24 hours have
elapsed since injury or if there is a risk of heavy contamination or following burns.
An intravenous preparation of tetanus antitoxin is appropriate for patients where large
doses are indicated (i.e., treatment of tetanus), or when the patient has a significant
haemostatic defect which may cause bleeding following intramuscular injection. In New
Zealand, Intragam® P is used to provide intravenous tetanus immunoglobulin. As the
level of immunoglobulin in each batch varies consultation with an NZBS Transfusion
Medicine Specialist/Medical Officer is recommended prior to prescription.
Treatment of Suspected or Confirmed Clinical Tetanus
For the treatment of suspected or confirmed clinical tetanus it is recommended that a
single dose Intragam® P containing 4000 IU tetanus immunoglobulin is given to cover
the typical 4-6 week course of the illness. NZBS acknowledges that the optimal dose,
which may be as low as 500 IU, is not yet defined.
Transfusion Medicine Handbook 3rd Edition
Page 83
5.4.5 Zoster Immunoglobulin-VF
Zoster Immunoglobulin-VF is a sterile, preservative-free solution containing not
less than 200 IU/vial varicella-zoster antibody. Plasma for Zoster Immunoglobulin-
VF is obtained from blood donors who have recently recovered from shingles or
chickenpox. Donations are selected on the basis that they contain high levels of
antibodies against Herpesvirus varicellae. Zoster Immunoglobulin-VF is intended for
intramuscular injection.
Indications for Use
Zoster Immunoglobulin-VF is indicated for prophylaxis against varicella in patients who
meet all of the following four criteria listed below.
Table 5.17: Clinical Indications for the Use of Zoster Immunoglobulin-VF
Criteria
Required Condition(s)
One of the following underlying
■
Neoplastic disease (leukaemia or lymphoma)
illnesses or conditions:
■
Congenital or acquired immune compromise
■
Immunosuppressive therapy with steroids or
antimetabolites
■
Pregnancy in non-immune woman
One of the following types of
■
Household contact
exposure to chickenpox or
■
Playmate contact (> 1 hour play indoors)
shingles patients:
■
Hospital contact (in same 2 to 4 bedroom or
adjacent beds in a large ward)
■
Newborn contact (newborn of mother who
develops chickenpox (but not zoster) from
7 days before to 7 days after delivery)
■
Premature infant ≥ 28 weeks gestation
whose mother lacks a prior history of
chickenpox
■
Premature infant < 28 weeks gestation or
< 1000 g regardless of maternal history
Negative or unknown prior history
of chickenpox
Zoster Immunoglobulin-VF can
be given within 96 hours1 for best
effect (however may be given up
to 10 days post-exposure)
1 For guidance on the management of both pregnant women exposed to varicella or zoster and newborns
exposed to maternal perinatal varicella or zoster, including recommendations on the timing of Zoster
Immunoglobulin-VF administration in these situations, refer to the NZ Ministry of Health Immunisation
Handbook.
Page 84
Transfusion Medicine Handbook 3rd Edition
The Starship Children’s Health Clinical Guideline covering
Zoster Immunoglobulin (2013)
and the National Child Cancer Network Guideline for
Immunisation of Children During
and After Cancer Therapy (2013) provide additional specific detail on the use of zoster
immunoglobulin in paediatric post-exposure prophylaxis and are summarised below.
Consultation with a specialist paediatrician or NZBS Transfusion Medicine Specialist/
Medical Officer is recommended prior to prescription.
Table 5.18: Recommendations for the Use of Zoster Immunoglobulin in Children at
Risk for Developing Serious Varicella Infection and Complications
At Risk Group
Comments
Considered as
High dose corticosteroid therapy
> 0.5 mg/kg/day of prednisone or
equivalent within the last three months.
Receiving immunosuppressive
All children regardless of VZV IgG status.
treatment or chemotherapy
For 3 months after therapy if VZV IgG negative.
Completed immunosuppressive
Regardless of VZV IgG status:
treatment or chemotherapy1
- for 6 months after autologous HPC transplant.
- for 12 months2 after allogeneic HPC transplant.
Malignancy
Congenital immunodeficiency
HIV positive; no prior history of
chickenpox
1 Patients who have received antithymocyte globulin, alemtuzumab, fludarabine or other T-cell
immunomodulation should be discussed with a paediatric oncologist.
2May be longer if chronic GVHD is present.
Varicella-zoster immunoglobulin is of no value in the treatment of established varicella
or zoster infection. High levels of circulating antibody do not prevent dissemination
of infection.
Dosage and Administration
The required dose is 125 IU per 10 kg bodyweight and rounded up to the nearest
200 IU to a maximum of 600 IU.
Table 5.19: Weight-Based Dosing Schedule for Zoster Immunoglobulin-VF
Weight of Patient (kg)
Dose (IU)
No. of Vials
0 - 10
125
1
10.1 - 20
250
2
20.1 - 30
375
2
30.1 - 40
500
3
>40
600
3
Transfusion Medicine Handbook 3rd Edition
Page 85
An intravenous preparation of varicella-zoster immunoglobulin is appropriate when
the patient has a significant haemostatic defect which may cause bleeding following
intramuscular injection. In New Zealand, Intragam® P is used to provide intravenous
varicella-zoster immunoglobulin. As the level of immunoglobulin in each batch
varies, consultation with an NZBS Transfusion Medicine Specialist/Medical Officer is
recommended prior to prescription.
5.4.6 Berirab® P (Rabies immunoglobulin)
Berirab® P does not have full New Zealand registration and so consultation with an
NZBS Transfusion Medicine Specialist/Medical Officer is required prior to product
release.
Berirab® P is a sterile solution of anti-rabies immunoglobulin prepared from the plasma
of individuals immunised with rabies vaccine. Donations are selected on the basis that
they contain high levels of rabies antibody. Each 1 mL of Berirab® P contains a minimum
of 150 IU/mL of rabies antibody. Berirab® P should be administered intramuscularly.
If anatomically feasible, the full dose should be thoroughly infiltrated in the area around
and into the wounds. Any remaining volume should be injected intramuscularly at a
site distant from vaccine administration.
Indications for Use
Berirab® P is indicated for prophylaxis in individuals suspected of having exposure
to rabies from:
■
Bites, licks, scratches or other injuries caused by a suspected rabid animal
■
Mucous membrane contamination with infectious tissue or saliva of suspected
rabid animal
■
Mucous membrane or new skin wound contact with rabies live attenuated
vaccine (e.g., vaccination baits)
The exception to these indications is individuals who have previously been immunised,
pre- or post-exposure, with rabies vaccine prepared from human diploid cells.
The recommended dose of Berirab® P is 20 IU per kg body weight which should be
given in conjunction with rabies vaccine.
5.4.7 Rh(D) Immunoglobulin-VF (Anti-D immunoglobulin)
Rh(D) Immunoglobulin-VF is a sterile, preservative-free solution with an anti-D antibody
content of either 625 IU or 250 IU per vial. Rh(D) Immunoglobulin-VF is prepared from
plasma obtained from either New Zealand voluntary or USA remunerated donors who
have been immunised to the RhD antigen. Rh(D) Immunoglobulin-VF is intended for
deep intramuscular injection using an appropriate sized needle.
Anti-D immunoglobulin acts by suppressing the immune response of RhD negative
individuals exposed to RhD positive red cells. Such exposure follows the passage of
cells from the fetal to the maternal circulation or the transfusion of RhD positive red
cells. Clinical studies indicate that the administration of anti-D immunoglobulin to a
RhD negative mother within 72 hours of the birth of a RhD positive infant reduces
Page 86
Transfusion Medicine Handbook 3rd Edition
the incidence of RhD isoimmunisation from 12 - 13% to 1 - 2%. Studies have also
shown that this number can be reduced to < 1.0% by antenatal prophylaxis with anti-D
immunoglobulin administered at 28 and 34 weeks of pregnancy.
Indications for Use
Rh(D) Immunoglobulin-VF is indicated for:
■
Prevention of RhD sensitisation in RhD negative females with child-bearing
potential
Rh(D) Immunoglobulin-VF may, in certain circumstances, also be used for protection
against the development of anti-D sensitisation when RhD positive donor red cells
are transfused to RhD negative females without child-bearing potential and to RhD
negative males.
Precautions
There is some evidence to suggest that intramuscular administration of anti-D
immunoglobulin may be associated with an increased risk of lack of effect in patients
with a body mass index (BMI) > 30. An Expert Panel Consensus Position Statement
(available online at www.transfusion.com.au/node/612) provides recommendations
regarding the use of anti-D immunoglobulin in these patients.
Contraindications
Rh(D) Immunoglobulin-VF should not be given to RhD positive individuals or to RhD
negative individuals previously sensitised to the RhD antigen.
Rh(D) Immunoglobulin-VF should not be given to RhD negative women with
detectable anti-D except where the antibody is passively acquired due to prior
antenatal administration. If unsure whether the anti-D detected in the mother’s blood
is passively acquired or preformed, the treating clinician and/or a NZBS Transfusion
Medicine Specialist/Medical Officer should be consulted. If there is continuing doubt,
Rh(D) Immunoglobulin-VF should be administered. Although there is no benefit in
administering Rh(D) Immunoglobulin-VF to a woman who is already sensitised to RhD
antigen, there is no more risk than when it is given to a woman who is not sensitised.
Dosage and Administration: Antenatal and Postpartum Prophylaxis
The following table is based on the 2003 Australian National Blood Authority (NBA)
Guidelines on the Prophylactic Use of RhD Immunoglobulin (Anti-D) in Obstetrics and
the 2013 British Committee for Standards in Haematology
Guideline for the Use of
Anti-D Immunoglobulin for the Prevention of HDFN. The table below is reproduced,
together with additional explanatory notes, in Section 6.8:
Hemolytic Disease of the
Fetus and Newborn (HDFN) of this Handbook. In 2011 the Royal Australian and New
Zealand College of Obstetricians and Gynaecologists (RANZCOG) endorsed the 2003
NBA
Guidelines, however it should be noted that the NZ Ministry of Health does not
recommend routine antenatal anti-D prophylaxis (RAADP).
Transfusion Medicine Handbook 3rd Edition
Page 87
Table 5.20: Indications for the Use of Anti-D Immunoglobulin for the Prevention of
HDFN (unless the fetus is confirmed to be RhD negative)
Timing
Clinical Indication
Routine antenatal
A RhD negative woman, not previously immunised
prophylaxis1
to produce anti-D; one dose at each of 28 and 34
weeks gestation.2
Anti-D immunoglobulin dose: 625 IU (125 µg)
Potential sensitising event A RhD negative woman, not previously immunised to
during first trimester up
produce anti-D, with an obstetric indication.
to and including 12 weeks ■ Uterine bleeding where this repeated, heavy or
gestation
associated with abdominal pain3
■
Miscarriage3
■
Termination of pregnancy
■
Ectopic pregnancy
■
Molar pregnancy
Anti-D immunoglobulin dose: 250 IU (50 µg)4
Potential sensitising
A RhD negative woman, not previously immunised to
event beyond first
produce anti-D, with an obstetric indication.
trimester5
■
Amniocentesis, chorionic villus sampling, and
intrauterine fetal blood sampling
■
Antepartum haemorrhage (or unexplained
uterine pain)
■
External cephalic version (performed or
attempted)
■
Abdominal trauma sufficient to cause FMH
■
Ectopic pregnancy
■
Molar pregnancy
■
Intrauterine death or stillbirth
■
Miscarriage, threatened miscarriage
■
Termination of pregnancy
Anti-D immunoglobulin dose: 625 IU (125 µg)
Postpartum prophylaxis6
A RhD negative woman, not previously immunised
to produce anti-D, who gives birth to a RhD positive
baby.
Anti-D immunoglobulin dose: 625 IU (125
µg) with additional dose(s) indicated where
fetomaternal haemorrhage is > 6 mL fetal red
cells
1 Routine antenatal anti-D prophylaxis (RAADP) should be administered regardless of, and in addition to,
prophylaxis given for a potentially sensitising event.
2 A sample for antibody testing should be taken prior to administration of anti-D immunoglobulin at 28 weeks.
3 Before 12 weeks gestation, in cases of either spontaneous complete miscarriage where the uterus is
not instrumented or mild painless vaginal bleeding, the risk of fetomaternal haemorrhage is negligible.
4 For multiple pregnancies the recommended anti-D immunoglobulin dose is 625 IU (125 µg).
5 Routine antenatal prophylaxis does not preclude prophylaxis for a potentially sensitising event.
6 Routine antenatal prophylaxis or prophylaxis for a potentially sensitising event does not preclude postpartum
prophylaxis.
Page 88
Transfusion Medicine Handbook 3rd Edition
NZBS is aware that an alternative approach to RAADP may be to administer a single
1500 IU anti-D dose at 28 weeks, however this is currently not endorsed in NZ.
Dosage and Administration: Transfusion of RhD Positive Blood Components
The recommended dose of Rh(D) Immunoglobulin-VF is 100 IU per mL RhD positive
red cells. A single 625 IU dose of Rh(D) Immunoglobulin-VF will therefore suppress the
immune response induced by up to 6 mL of RhD positive red cells. Consultation with
a NZBS Transfusion Medicine Specialist/Medical Officer is recommended in the event
that a recipient has been transfused with a larger volume of RhD positive red cells.
The amount of red cell contamination in platelet components supplied by NZBS is less
than 1 mL per unit and therefore adequately covered by a single 250 IU dose of Rh(D)
Immunoglobulin-VF. A single 250 IU dose of Rh(D) Immunoglobulin-VF (administered
subcutaneously in severely thrombocytopenic patients) is likely to be sufficient to cover
up to five adult therapeutic doses of RhD positive platelets given within a 6-week period.
5.4.8 Rhophylac® (Anti-D immunoglobulin)
Rhophylac does not have full New Zealand registration and so consultation with an
NZBS Transfusion Medicine Specialist is required prior to product release.
Rhophylac® is a sterile, preservative-free solution in a pre-filled 2 mL syringe containing
1500 IU (300 µg) anti-D IgG immunoglobulin. Rhophylac® is manufactured from plasma
obtained from remunerated donors who have been immunised to the RhD antigen. It is
important to note that this product is obtained from a screened and carefully monitored
donor pool and has a similar safety profile to Rh(D) Immunoglobulin-VF. Rhophylac®
may safely be administered intramuscularly or intravenously.
Indications for Use
Rhophylac® has similar indications and contraindications for use as other anti-D
immunoglobulin products. While Rhophylac® is an acceptable alternative to Rh(D)
Immunoglobulin-VF, it is supplied by NZBS only when stock of this product is unavailable,
large doses of anti-D are indicated (i.e., > 2 vials 625 IU Rh(D) Immunoglobulin-VF), or
when a product suitable for intravenous administration is required.
Dosage and Administration
A maximum dose of 15,000 IU is sufficient in the case of larger incompatible
transfusions independent of whether the transfusion volume is greater than
300 mL of RhD positive red cells. Treatment can usually be given without
preceding exchange transfusion when the transfused RhD positive blood represents
less than 20% of the total circulating red blood cells. If the volume exceeds 20%,
consideration should be given to red cell exchange transfusion to reduce the load of
RhD positive cells prior to Rhophylac® administration.
Precautions
■
Following a large fetomaternal bleed or incompatible transfusion, patients
receiving large doses of anti-D immunoglobulin should be monitored, as there
is a risk of haemolytic reaction. To reduce this risk, the maximum recommended
dose administration rate is 3000 IU every 8 hours.
■
Rhophylac® can contain antibodies to other Rh antigens and the passive transfer
Transfusion Medicine Handbook 3rd Edition
Page 89
of these antibodies may be detectable by serological testing methods.
■
There is some evidence to suggest that intramuscular administration of anti-D
immunoglobulin may be associated with an increased risk of lack of effect
in patients with a body mass index (BMI) > 30. An Expert Panel Consensus
Position Statement (available online at www.transfusion.com.au/node/612)
provides recommendations regarding the use of anti-D immunoglobulin in these
patients.
5.4.9 Intragam® P (Normal immunoglobulin, intravenous, IVIg)
Intragam® P is a sterile, preservative free solution containing 6 g of human protein and
10 g of maltose in each 100 mL, available in 10 mL (0.6 g), 50 mL (3 g) and 200 mL
(12 g) vials. The solution has a pH of 4.25 and isotonicity is achieved by the addition
of maltose. Intragam® P is made by chromatographic fractionation of large pools of
human plasma obtained from New Zealand’s voluntary and non-remunerated blood
donors. Intragam® P is intended for intravenous administration.
The distribution of IgG subclasses present in Intragam® P is: IgG1 (61%), IgG2 (36%),
IgG3 (3%) and IgG4 (1%). Intragam® P contains only trace amounts of IgA (typically
<0.025 mg/mL). The actual amount of IgA in each batch is printed on the label.
The protein has not been chemically or enzymatically modified and the manufacturing
process contains specific steps to reduce the possibility of virus transmission including
pasteurisation (heating at 60°C for 10 hours) and incubation at low pH.
Indications for Use
Intragam® P is registered for use as replacement IgG therapy and immunomodulatory
therapy for a number of conditions.
Table 5.21: Registered Indications for Use of Intragam® P
Indication
Comments
Replacement Therapy
■
Primary immunodeficiency
Causing hypogammaglobulinaemia
■
Acquired hypogammaglobulinaemia
Symptomatic with recurrent infections
Immunomodulatory Therapy
■
Primary immune thrombocytopaenia1
■
Kawasaki disease
■
Guillain-Barre syndrome
1 Recommendations for the use of IVIg in immune thrombocytopaenia (ITP) can be found in the International
Consensus Report on the Investigation and Management of Primary Immune Thrombocytopenia in Blood
2010; 115: 168-186 and the American Society of Hematology (ASH) 2011 Clinical Practice Guideline on
the Evaluation and Management of Immune Thrombocytopenia.
In addition, intravenous immunoglobulin has an established and emerging therapeutic
role in a wide range of, and in some cases uncommon or rare, autoimmune and
inflammatory diseases including chronic inflammatory demyelinating polyneuropathy,
Page 90
Transfusion Medicine Handbook 3rd Edition
inflammatory myopathies, Lambert–Eaton myasthenic syndrome, multifocal motor
neuropathy, myasthenia gravis, and stiff-person syndrome. Intragam® P is not
registered for these indications in New Zealand and issue of this product is subject
to consultation between the specialist physician and a NZBS Transfusion Medicine
Specialist/Medical Officer.
Comprehensive evidence-based guidelines for the use of IVIg are lacking. Substitutes,
such as the 2012 Australian National Blood Authority
Criteria for the Clinical Use of
Intravenous Immunoglobulin in Australia (2nd ed.) and the 2011 UK Department of
Health
Clinical Guidelines for Immunoglobulin Use (2nd ed. update) provide information
about the criteria for accessing IVIg and should be followed wherever possible to avoid
the inappropriate utilisation of Intragam® P.
Dosage and Administration
Intragam® P may be infused undiluted. It may also be infused diluted with up to 2 parts
of 0.9% saline or 5% glucose.
■
The infusion should be commenced at the rate of 1 mL per minute.
■
After 15 minutes the rate may be gradually increased to a maximum of 3 - 4
mL per minute over a further 15 minutes.
■
Too rapid a rate of infusion may cause flushing and changes in heart rate and
blood pressure.
■
Patients naive to Intragam® P, switching from an alternative IVIg product, or who
have not received IVIg for a long time, should be closely monitored during the
first infusion.
■
In patients at risk for acute renal failure or thromboembolic adverse reactions,
IVIg products should be administered at the minimum rate of infusion and dose
practicable.
Replacement Therapy
■
The optimal dose and frequency of administration of Intragam® P must be
determined for each patient. Freedom from recurrent bacterial infections is
usually achieved with a serum IgG level > 5 g/L.
■
Most patients initially receive 400 mg IgG per kilogram body weight, followed
by monthly maintenance doses of at least 200 mg per kilogram body weight.
■
The monthly maintenance dose, guided by the patient’s clinical status and
pre-infusion (trough) serum IgG level, is often 300 - 450 mg of IgG per kilogram
body weight.
■
As catabolic rates vary, the IgG levels of new patients should be monitored
regularly for several monthly cycles to determine the effective dose.
Immunomodulatory Therapy
Primary immune thrombocytopaenia
■
Patients should receive up to a maximum total cumulative dose of 2.0 g IgG
per kilogram body weight, over 2 - 5 days.
Transfusion Medicine Handbook 3rd Edition
Page 91
Kawasaki disease
■
Patients should receive 1.6 - 2.0 g IgG per kilogram body weight, administered
in divided doses over 2 - 5 days, or 2.0 g IgG per kilogram body weight as a
single dose.
Guillain-Barre syndrome
■
Patients should receive 0.4 g IgG per kilogram body weight per day for five days.
IVIg dosage recommendations for off-label indications are available in
Criteria for the
Clinical Use of Intravenous Immunoglobulin in Australia (2nd ed.), developed by the
Australian National Blood Authority (NBA) in 2012.
Contraindications
Intragam® P is contraindicated in individuals who have had a true anaphylactic reaction
to the active substance or the excipient.
Precautions
■
Administration
Intragam® P should only be administered intravenously. It is possible that
Intragam® P may, on rare occasions, cause a precipitous fall in blood pressure
and a clinical picture of anaphylaxis. Therefore, adrenaline and oxygen should
be available for the treatment of such an acute reaction.
■
Aseptic meningitis
Aseptic meningitis syndrome has been reported to occur infrequently in
association with IVIg treatment.
■
IgA antibodies
Intragam® P contains trace amounts of IgA which may provoke anaphylaxis in
patients with IgA antibodies, such as those with IgA deficiency.
■
Positive direct antiglobulin tests and red cell haemolysis
Positive direct antiglobulin tests and red cell haemolysis have been reported
following high dose infusion of intravenous immunoglobulin due to the presence
of anti-A, anti-B, and occasionally with anti-D or other erythrocyte antibodies
in the product. Such red cell sensitisation may cause crossmatching difficulties
and transient haemolytic anaemia.
■
Renal dysfunction
There have been reports of renal dysfunction and acute renal failure in patients
receiving IVIg. Patients should be adequately hydrated prior to administration of IVIg.
■
Thromboemblism
Thrombotic events have been reported in association with IVIg therapy. Caution
should be exercised in prescribing and administering Intragam® P in in patients
with pre-existing risk factors for thrombotic events.
■
Thrombophlebitis
Prolonged administration (over 6 hours) using large doses (greater than 400
mg/kg) may result in thrombophlebitis at the infusion site.
Page 92
Transfusion Medicine Handbook 3rd Edition
Adverse Reactions
Reactions to intravenous immunoglobulin tend to be related to the infusion rate and
are most likely to occur during the first hour of the infusion. It is recommended that the
patient’s vital signs and general status be monitored regularly throughout the infusion.
The types of reactions that may occur include: malaise, abdominal pain, headache,
chest-tightness, facial flushing or pallor, hot sensations, dyspnoea, non-urticarial skin
rash, itching, arthralgia, tissue swelling, hypotension, nausea, or vomiting. Should
any of these reactions develop during infusion of Intragam® P, the infusion should
be temporarily stopped (5 - 10 minutes) until the patient improves clinically and then
cautiously recommenced at a slower rate.
Allergic reactions are most likely to occur during the first hour of the infusion.
True hypersensitivity reactions to intravenous immunoglobulin such as urticaria,
angioedema, bronchospasm or hypotension occur very rarely. Should an anaphylactic
reaction to Intragam® P develop, the infusion should be stopped and immediate
treatment instituted with adrenaline and oxygen.
Interactions with Other Medicines
Passively acquired antibody can interfere with the response to live attenuated virus
vaccines such that vaccine administration should be deferred for at least 3 months.
In the case of measles and varicella vaccines following IVIg products, the impairment
may persist for up to 12 months. Where deferral is impractical, patients receiving
such vaccines should have their antibody response checked. By the same token,
immunoglobulins should not be administered for at least two weeks after live attenuated
vaccines are given. Consultation with a NZBS Transfusion Medicine Specialist/Medical
Officer is recommended.
Interaction with Capillary Glucose Measurement
Caution should be exercised when interpreting blood sugar levels in patients receiving
Intragam® P. The maltose present in Intragam® P may result in falsely elevated capillary
blood glucose levels with some types of glucose meters. If this measurement is used
to guide treatment, hypoglycaemia may occur.
When monitoring glucose levels in patients receiving Intragam® P, consult the product
information and/or manufacturer of the glucose meter and test strips (including those
used at home by patients) to ensure that maltose does not interfere with the blood
glucose reading.
5.4.10 Privigen® (Normal immunoglobulin, intravenous, IVIg)
Privigen® is a sterile, preservative free 10% solution containing 10 g of human protein
in each 100 mL, available in 50 mL (5 g), 100 mL (10 g) and 200 mL (20 g) vials. The
solution has a pH of 4.8 and is approximately isotonic. The product contains 250
mmol/L of L-proline as a stabiliser which is a physiological non-essential amino acid.
It contains no carbohydrate stabiliser (e.g., sucrose, maltose) and has a low sodium
content. Privigen® is made by a combination of cold ethanol fractionation, octanoic
acid fractionation and anion exchange chromatography of large pools of human plasma
obtained from blood donors in the United States and Europe. Privigen® is intended
for intravenous administration.
Transfusion Medicine Handbook 3rd Edition
Page 93
The distribution of IgG subclasses present in Privigen® is: IgG1 (68%), IgG2 (29%),
IgG3 (2%) and IgG4 (1%). Privigen® contains only trace amounts of IgA (typically
<0.025 mg/mL).
The protein has not been chemically or enzymatically modified and the manufacturing
process contains specific steps to reduce the possibility of virus transmission including
filtration and incubation at low pH.
Indications for Use
Privigen® is registered for use as replacement IgG therapy and immunomodulatory
therapy for a number of conditions.
Table 5.22: Registered Indications for Use of Privigen®
Indication
Comments
Replacement Therapy
■
Primary immunodeficiency1
Causing hypogammaglobulinaemia
■
Acquired hypogammaglobulinaemia
Symptomatic with recurrent infections
Immunomodulatory Therapy2
■
Primary immune thrombocytopaenia3, 4
■
Kawasaki disease
■
Guillain-Barré syndrome
■
Chronic inflammatory demyelinating
polyneuropathy
■
Multifocal motor neuropathy
■
Myasthenia gravis exacerbations
■
Lambert-Eaton myasthenic syndrome
■
Stiff person syndrome
1 The use of Privigen® has not been established in patients with primary immunodeficiency disorder under
the age of 3 years.
2 The use of Privigen® has not been established in patients with neurological indications under the age of
18 years.
3 Recommendations for the use of IVIg in immune thrombocytopaenia (ITP) can be found in the International
Consensus Report on the Investigation and Management of Primary Immune Thrombocytopenia in Blood
2010; 115: 168-186 and the American Society of Hematology (ASH) 2011 Clinical Practice Guideline on
the Evaluation and Management of Immune Thrombocytopenia.
4 The use of Privigen® has not been established in patients with ITP under the age of 15 years.
In addition, intravenous immunoglobulin has an established and emerging therapeutic
role in a wide range of other, and in some cases also uncommon or rare, autoimmune
and inflammatory diseases. Privigen® is not registered in New Zealand for indications
other than those listed. For situations involving off-label indications, issue of this product
is subject to consultation between the specialist physician and a NZBS Transfusion
Medicine Specialist/Medical Officer.
Comprehensive evidence-based guidelines for the use of IVIg are lacking. Substitutes,
Page 94
Transfusion Medicine Handbook 3rd Edition
such as the 2012 Australian National Blood Authority
Criteria for the Clinical Use of
Intravenous Immunoglobulin in Australia (2nd ed.) and the 2011 UK Department of
Health
Clinical Guidelines for Immunoglobulin Use (2nd ed. update) provide information
about the criteria for accessing IVIg and should be followed wherever possible to avoid
the inappropriate utilisation of Privigen®.
Dosage and Administration
Privigen® may be infused undiluted. It may also be infused diluted with 5% glucose.
■
Patients naive to Privigen®, switching from an alternative IVIg product, or who
have not received IVIg for a long time, should be closely monitored during and
for the first hour after the first infusion.
■
In such patients, the infusion should be commenced at the rate of 0.3 mL per
kilogram body weight per hour.
■
If well tolerated, the rate may gradually be increased to 4.8 mL per kilogram
body weight per hour.
■
In patients at risk for acute renal failure or thromboembolic adverse reactions,
IVIg products should be administered at the minimum rate of infusion and dose
practicable.
Replacement Therapy
■
The optimal dose and frequency of administration of Privigen® must be
determined for each patient.
■
Most patients receive 400 mg IgG per kilogram body weight initially, followed
by monthly maintenance doses of at least 200 mg per kilogram body weight.
■
The monthly maintenance dose, guided by the patient’s clinical status, is often
300 - 450 mg of IgG per kilogram body weight aiming for a pre-infusion (trough)
serum IgG level of at least 4 - 6 g/L.
■
As catabolic rates vary, the IgG levels of new patients should be monitored
regularly for several monthly cycles to determine the effective dose. Three to
six months are required for equilibration.
Immunomodulatory Therapy
Primary immune thrombocytopaenia
■
Patients should receive up to a maximum total cumulative dose of 2.0 g IgG
per kilogram body weight, over 2 - 5 days.
Kawasaki disease
■
Patients should receive 1.6 - 2.0 g IgG per kilogram body weight, administered
in divided doses over 2 - 5 days, or 2.0 g IgG per kilogram body weight as a
single dose.
Transfusion Medicine Handbook 3rd Edition
Page 95
Guillain-Barre Syndrome
■
Patients should receive 0.4 g IgG per kilogram body weight per day for five
days.
Chronic inflammatory demyelinating polyneuropathy
■
Patients should receive a starting dose of 2.0 g IgG per kilogram body weight,
administered in divided doses over 2 - 5 days.
■
Patients should receive a maintenance dose of 1.0 g IgG per kilogram body
weight, administered every three weeks.
Multifocal motor neuropathy
■
Patients should receive a starting dose of 2.0 g IgG per kilogram body weight,
administered in divided doses over 2 - 5 days.
■
Patients should receive a maintenance dose of 0.4 - 2.0 g IgG per kilogram
body weight, administered every two to six weeks.
Myasthenia gravis exacerbations
■
Prior to surgery or during myasthenic crisis, patients should receive an induction
dose of 1.0 - 2.0 g IgG per kilogram body weight, administered in divided doses
over 2 - 5 days.
■
Patients should receive a maintenance dose of 0.4 - 1.0 g IgG per kilogram
body weight, administered every four to six weeks.
Lambert-Eaton myasthenic syndrome
■
Patients should receive a starting dose of 2.0 g IgG per kilogram body weight,
administered in divided doses over 2 - 5 days.
■
Patients should receive a maintenance dose of 0.4 - 1.0 g IgG per kilogram
body weight, administered every two to six weeks.
Stiff person syndrome
■
Patients should receive a starting dose of 2.0 g IgG per kilogram body weight,
administered in divided doses over 2 - 5 days.
■
Patients should receive a maintenance dose of 1.0 - 2.0 g IgG per kilogram
body weight, administered every four to six weeks.
IVIg dosage recommendations for off-label indications are available in
Criteria for
the Clinical Use of Intravenous Immunoglobulin in Australia (2nd ed.), developed
by the Australian National Blood Authority (NBA) in 2012. In the case of allogeneic
haematopoietic stem cell transplant, the following dosage recommendations for
Privigen® have been used.
Treatment of infections and prophylaxis of graft-versus-host disease
■
Patients should receive 0.5 g IgG per kilogram body weight weekly from 7 days
before to 3 months after transplantation.
Persistent hypogammaglobulinaemia
■
Patients should receive 0.5 g IgG per kilogram body weight monthly until antibody
levels return to normal.
Page 96
Transfusion Medicine Handbook 3rd Edition
Contraindications
Privigen® is contraindicated in individuals who have had a true anaphylactic reaction
to the active substance or the excipient and in those with hyperprolinaemia.
Precautions
■
Administration
Privigen® should only be administered intravenously. IVIg may, on rare occasions,
cause a precipitous fall in blood pressure and a clinical picture of anaphylaxis.
Therefore, adrenaline and oxygen should be available for the treatment of such
an acute reaction.
■
Aseptic meningitis
Aseptic meningitis syndrome has been reported to occur infrequently in
association with IVIg treatment.
■
IgA antibodies
Privigen® contains trace amounts of IgA which may provoke anaphylaxis in
patients with IgA antibodies, such as those with IgA deficiency.
■
Positive direct antiglobulin tests and red cell haemolysis
Positive direct antiglobulin tests and red cell haemolysis have been reported
following high dose infusion of intravenous immunoglobulin due to the presence
of anti-A, anti-B, and occasionally with anti-D or other erythrocyte antibodies
in the product. Such red cell sensitisation may cause crossmatching difficulties
and transient haemolytic anaemia.
■
Renal dysfunction
There have been reports of renal dysfunction and acute renal failure in patients
receiving IVIg. Patients should be adequately hydrated prior to administration of IVIg.
■
Thromboembolism
Thrombotic events have been reported in association with IVIg therapy. Caution
should be exercised in prescribing and administering Privigen® in patients with
pre-existing risk factors for thrombotic events.
Adverse Reactions
Reactions to intravenous immunoglobulin tend to be related to the infusion rate and
are most likely to occur during the first hour of the infusion. It is recommended that the
patient’s vital signs and general status be monitored regularly throughout the infusion.
The types of reactions that may occur include: malaise, abdominal pain, headache,
chest-tightness, facial flushing or pallor, hot sensations, dyspnoea, non-urticarial
skin rash, itching, arthralgia, tissue swelling, hypotension, nausea, or vomiting.
Should any of these reactions develop during infusion of Privigen®, the infusion should
be temporarily stopped (5-10 minutes) until the patient improves clinically and then
cautiously recommenced at a slower rate.
Allergic reactions are most likely to occur during the first hour of the infusion.
True hypersensitivity reactions to intravenous immunoglobulin such as urticaria,
angioedema, bronchospasm or hypotension occur very rarely. Should an anaphylactic
reaction to Privigen® develop, the infusion should be stopped and immediate treatment
instituted with adrenaline and oxygen.
Transfusion Medicine Handbook 3rd Edition
Page 97
Interactions with Other Medicines
Passively acquired antibody can interfere with the response to live attenuated virus
vaccines such that vaccine administration should be deferred for at least 3 months.
In the case of measles and varicella vaccines following IVIg products, the impairment
may persist for up to 12 months. Where deferral is impractical, patients receiving
such vaccines should have their antibody response checked. By the same token,
immunoglobulins should not be administered for at least two weeks after live attenuated
vaccines are given. Consultation with a NZBS Transfusion Medicine Specialist/Medical
Officer is recommended.
5.4.11 Evogam® (Normal immunoglobulin, subcutaneous, SCIg)
Evogam® is a sterile, preservative free solution containing 16 g of human protein in
each 100 mL, available in 5 mL (0.8 g) and 20 mL (3.2 g) vials. The solution has a
pH of 6.6. As a stabiliser, Evogam® contains 2.25g of glycine in each 100mL. It does
not contain a carbohydrate stabiliser (e.g., sucrose, maltose). Evogam® is made by
chromatographic fractionation of large pools of human plasma obtained from New
Zealand’s voluntary and non-remunerated blood donors. Evogam® is intended for
subcutaneous administration.
The distribution of IgG subclasses present in Evogam® is: IgG1 (48-58%), IgG2
(39-49%), IgG3 (1-2%) and IgG4 (1-2%). Evogam® contains only trace amounts
of IgA (typically <0.025 mg/mL). The actual amount of IgA in each batch is printed
on the label.
The protein has not been chemically or enzymatically modified and the manufacturing
process contains specific steps to reduce the possibility of virus transmission including
pasteurisation (heating at 60°C for 10 hours) and nanofiltration.
Indications for Use
Evogam® is indicated in adults and children for replacement therapy in:
■
Primary immunodeficiency diseases (PID)
■
Symptomatic hypogammaglobulinaemia secondary to underlying disease or
treatment
Dosage and Administration
The dose and dosage interval must be individualised for each patient based on their
measured IgG trough levels and ongoing clinical response. A weekly dose in the range
0.05 - 0.15 g/kg body weight is recommended, corresponding to a total monthly dose
of Evogam® in the range of 0.2 - 0.6 g/kg body weight.
Evogam® should be brought to room temperature and administered via the
subcutaneous route, optionally at several infusion sites (advisable for large > 20 mL
doses) and preferentially into the upper outer arm, upper thigh, abdomen or lateral hip
at an initial infusion rate of 10 mL/hour. The infusion rate may be gradually increased
after the first completed infusion up to 20 mL/hour. The maximum rate administered
during clinical trials was 40 mL/hour using two infusion pumps simultaneously. Evogam®
must not be administered intravenously.
Page 98
Transfusion Medicine Handbook 3rd Edition
Contraindications
Evogam® is contraindicated in individuals who have had a true anaphylactic reaction
to the active substance or to the excipient glycine.
Precautions
■
Administration
Evogam® must only be administered subcutaneously. Other routes of
administration have not been evaluated. Evogam® administered intravenously
could cause a clinical picture of anaphylaxis.
■
Aseptic meningitis
Aseptic meningitis syndrome has been reported to occur infrequently in
association with human immunoglobulin treatment.
■
IgA antibodies
Evogam® contains trace amounts of IgA which may provoke anaphylaxis in
patients with IgA antibodies, such as those with IgA deficiency.
■
Positive direct antiglobulin tests and red cell haemolysis
Evogam® can contain blood group antibodies causing a positive direct
antiglobulin tests and rarely red cell haemolysis. Evogam® recipients should be
monitored for clinical signs and symptoms of haemolysis.
■
Renal dysfunction
There have been occasional reports of renal dysfunction and acute renal failure
in patients receiving IVIg. In cases of renal impairment with Evogam® use,
discontinuation should be considered.
■
Thromboemblism
Thrombotic events have been reported in association with human immunoglobulin
therapy. Caution should be exercised in prescribing and administering Evogam®
in patients with pre-existing risk factors for thrombotic events.
Adverse Reactions
Local tolerability reactions of a mild to moderate intensity including infusion site pain,
injection site haematoma or pruritis, erythema, local heat and/or induration are very
commonly (≥1/10) reported at 8 to 12 hours after infusion. At 72 hours after infusion,
the frequency of reported symptoms markedly decreases and the incidence of local
reactions reduces with continued use of Evogam®. Other very commonly (≥ 1/10)
reported reactions include headache, fever, nausea, diarrhoea and vomiting. Less
common reactions include chills, back pain, arthralgia and hypotension.
Rarely, human immunoglobulin may cause allergic reactions and, in isolated cases,
anaphylactic shock. Should an anaphylactic reaction to Evogam® develop, the infusion
should be stopped and immediate treatment instituted with adrenaline and oxygen.
Interactions with Other Medicines
Passively acquired antibody can interfere with the response to live attenuated virus
vaccines such that vaccine administration should be deferred for at least 3 months. In
the case of measles and varicella vaccines following normal immunoglobulin products,
the impairment may persist for up to 12 months. Where deferral is impractical, patients
Transfusion Medicine Handbook 3rd Edition
Page 99
receiving such vaccines should have their antibody response checked. By the same
token, immunoglobulins should not be administered for at least two weeks after
live attenuated vaccines are given. Consultation with a NZBS Transfusion Medicine
Specialist/Medical Officer is recommended.
5.5
Other Products
5.5.1 Berinert® P (C1-esterase inhibitor)
Berinert® P does not have full New Zealand registration and so consultation with a NZBS
Transfusion Medicine Specialist/Medical Officer is required prior to release of this product.
Berinert® P is a C1-esterase inhibitor concentrate supplied as 500 IU per vial (50 IU/
mL). Berinert® P is intended for slow intravenous injection or infusion.
Indications for Use
Berinert® P is indicated for:
■
The management of patients with C1-esterase inhibitor deficiency and/or
hereditary angioedema (HAE)
Treatment should be initiated under the supervision of a physician experienced in the
management of C1-esterase inhibitor deficiency.
The treatment of capillary leak syndrome with Berinert® P is not advised.
Dosage and Administration
The recommended dose is 20 IU per kilogram body weight, rounded to the nearest
500 IU. An additional dose may be required in less than 5% of patients with persistent
or worsening clinical condition within a few hours of administration of the initial dose.
It is recommended that Berinert® P be administered by slow intravenous injection
4 mL/minute.
5.5.2 Products from Australian Red Cross Blood Service (ARCBS)
From time to time there may be occasions when fractionated products prepared
specifically from New Zealand plasma are unavailable or in short supply.
To ensure continuity of supply, NZBS may purchase supplementary stock of similar
products from ARCBS. These products, manufactured by CSL Behring (using plasma
from Australian donors) may be supplied in place of the equivalent New Zealand
product. As they are unapproved medicines in New Zealand, a supply application is
required under Section 29 of the Medicines Act 1981.
Page 100
Transfusion Medicine Handbook 3rd Edition
6
SPECIAL CIRCUMSTANCES
6.1
Management of Acute Blood Loss
Complications of major blood loss and massive transfusion associated with, for
example, trauma, burns, surgery, obstetric haemorrhage and major gastrointestinal
bleeding may jeopardise patients and challenge laboratory and blood transfusion
resources. A successful outcome requires prompt action and good communication
between clinical specialties, diagnostic laboratories, blood banks and NZBS.
Massive blood loss is usually defined as the loss of one blood volume within a 24 hour
period (equivalent to 7% of ideal body weight in adults; 8 - 9% in children). Alternative
definitions include the loss of 50% of blood volume within 3 hours and a rate of blood
loss of 150 mL/minute. It is important that major blood loss is recognised early and
appropriate action taken to prevent shock and its consequences.
The aim of treatment is the rapid and effective restoration of an adequate blood volume
and to maintain blood composition within safe limits with regard to haemostasis,
oxygen carrying capacity and biochemistry.
The essential features of management are:
■
Restoring blood volume to maintain tissue perfusion and oxygenation.
■
Achieving haemostasis by:
-
surgical control of bleeding.
-
correcting coagulopathy by expedient use of blood component therapy,
based on results from early laboratory haemostasis screening.
The following table indicates likely crystalloid and blood transfusion requirements in
response to acute blood loss, based on estimation of lost circulating volume.
Table 6.1: Transfusion Requirements in Response to Loss of Blood Volume
% Loss of Blood Volume
Action
15%
No need for transfusion unless blood loss is
(750 mL in an adult)
superimposed on pre-existing anaemia or when the
patient is unable to compensate for this quantity of
blood loss because of severe cardiac or respiratory
disease.
15-30%
Transfuse crystalloids. A requirement for red cell
(800-1500 mL in an adult)
transfusion is unlikely unless the patient has a pre-
existing anaemia, reduced cardiopulmonary reserve
or if blood loss continues.
30-40%
Rapid volume replacement required with crystalloids.
(1500-2000 mL in an adult)
Red cell transfusion will probably be required.
>40%
Rapid volume replacement is required including red
(>2000 mL in an adult)
cell transfusion.
Transfusion Medicine Handbook 3rd Edition
Page 101
If bleeding continues after attempted surgical haemostasis and when the coagulation
tests are abnormal or the platelet count reduced, then platelets, fresh frozen plasma,
cryoprecipitate or a combination of these products may also be required.
In the setting of trauma-induced bleeding, early initiation of blood transfusion
support with optimal ratios of plasma and platelets to red cell units may help achieve
haemostasis and reduce the risk of exsanguination.
In trauma patients, use of the antifibrinolytic tranexamic acid is considered standard
of care as an adjunct in arresting bleeding and should be administered as early as
possible and within three hours of the trauma. The CRASH-2 trial included over 20,000
trauma patients, at least 16 years old, with significant haemorrhage (or at risk of) who
were within 8 hours of initial injury. Compared to placebo, administration of tranexamic
acid 1g loading dose over 10 minutes followed by 1g infusion over 8 hours reduced
hospital mortality and death due to haemorrhage within 4 weeks of injury. No increase
in the rate of vascular occlusion (myocardial infarction, stroke, pulmonary embolism)
was seen with the use of tranexamic acid. For further information on the use of this
antifibrinolytic agent see Section 8.7:
Tranexamic Acid.
Clinical trials in humans have not demonstrated albumin solutions or other colloids
to be superior to crystalloid in resuscitation, but larger quantities of crystalloid may
be required. Synthetic colloids such as dextrans and hydroxyethyl starch should be
avoided in patients at risk for acute kidney injury and otherwise limited to 1.5 litres
per 24 hours in adults.
Aggressive volume resuscitation may cause problems with interstitial oedema,
compartment syndrome, acute lung injury and, subsequent to haemodilution,
exacerbations of anaemia, thrombocytopaenia and coagulopathy. A strategy of
permissive hypotension, with minimal volume resuscitation and tolerating systolic
blood pressures of 80-100 mmHg, is generally preferable while active bleeding is
being controlled. Permissive hypotension is contraindicated in patients with traumatic
brain injury and should be used with caution in the elderly.
Large quantities of saline may cause hyperchloraemic metabolic acidosis with
subsequent complications and this has increasingly led to the use of physiologically
buffered fluids such as Plasmalyte 148 and compound sodium lactate (Hartmann’s
or Ringer-Lactate).
Avoid saline in patients with severe liver disease for whom sodium overload is a risk.
Specialist advice is recommended. For the same reason care should be taken with
Albumex® 4 in these patients.
Table 6.2:
Transfusion Support for Major Bleeding should be referred to in conjunction
with a local massive transfusion protocol (MTP).
Page 102
Transfusion Medicine Handbook 3rd Edition
esults
esponsibility
eat risk for a
esults available
esuscitation of trauma
ed cell transfusion
estimated
elated adverse outcome
ed as soon as possible and within 3
fected by colloid infusion
e FBC and coagulation r
, befor
rong blood in tube (WBIT) poses gr
Blood loss, including concealed blood loss, is
often under
Refer to local guidelines on r
patients and use of r
Monitor CVP if haemodynamically unstable
A named senior person should take r
for communication and documentation
Following trauma, tranexamic acid should be
administer
hours
Take samples at earliest opportunity as r
may be af
W
transfusion-r
May need to give FFP and platelets, as per local
MTP
Comments
■
■
■
■
■
■
■
■
>30
e-warmed
d blood volume
, fibrinogen at least
es
ofile, arterial blood gas
, fibrinogen, Blood Bank
ocedur
e peripheral cannulae
opriate surgical team
ect identity for transfusion samples
opriate, tranexamic acid, as a 1g loading
e corr
Insert wide bor
Give adequate volumes of pr
crystalloid +/- colloid
Aim to maintain normal BP and urine output
mL/hr in adults (or 0.5 mL/kg/hour)
Most appr
Duty anaesthetist
Blood Bank
Early surgical or obstetric intervention
Upper GI tract pr
Interventional radiology
If appr
dose over 10 minutes, followed by infusion of 1g
over 8 hours
FBC, PT/INR, APTT
sample, biochemical pr
Ensur
Repeat FBC, PT/INR, APTT
every 4 hours, or after one thir
replacement, or after FFP
Intervention
■
■
■
■
■
■
■
■
■
■
■
■
■
, it may
estrict volume
diac injury
ransfusion Support for Major Bleeding
culating volume
e cir
opriate to r
est bleeding
Table 6.2: T
Activity
Restor
Note: in patients with major
vessel or car
be appr
replacement after discussion with
surgical team
Contact key personnel
Arr
Request laboratory
investigations
Transfusion Medicine Handbook 3rd Edition
Page 103
e
e
e using
e
esults
e laboratory
elates with
een befor
/L for multiple/9
, fibrinogen befor
ol corr
e laboratory r
oup and scr
≥ 100 x 10
> 1.13 mmol/L2+
ovascular bleeding
ovascular bleeding
e available, however take FBC sample
> 1.5 x mean contr
fuse micr
eased micr
Contact Blood Bank or on-call scientist
Collect sample for gr
emergency stock
Emergency use of RhD positive blood is
acceptable if patient is male or post menopausal
female
Blood warmer indicated if large volumes ar
transfused rapidly
Consider use of cell salvage
Target platelet count
central nervous system trauma or with sever
dif
May need to transfuse platelets befor
results ar
first
PT/APTT
incr
Take sample for PT/INR, APTT
FFP transfused
May need to use FFP befor
available
Maintain ionised Ca
Comments
■
■
■
■
■
■
■
■
■
■
■
ol
/L after 2 x
/L
9
ovided following
9
e or 4 units for
oup specific will be
< 50 x 10
< 1.5 x mean contr
> 70 g/L
oup known (15 to 45
etransfusion testing
> 50 x 10
oximately 1 litr
eceipt of sample in laboratory)
outine pr
eplacement
oup O RhD negative
om r
ossmatched ABO gr
ovided when blood gr
ossmatch compatible units pr
Maintain haemoglobin
Blood needed immediately - use ‘emergency
stock’ gr
Blood needed in 15 to 45 minutes -
uncr
pr
minutes fr
Blood needed in 45 minutes or longer –
cr
completion of r
Maintain platelet count
Anticipate platelet count
blood volume r
Dose: 10 mL/kg for a neonate or small child,
otherwise one adult therapeutic dose
Anticipate coagulation factor deficiency after
blood loss of 1.5 x blood volume
Aim for PT/INR and APTT
Allow for 30 minutes thawing time
Dose: 12-15 mL/kg
(equivalent to appr
an average 70 kg adult)
Intervention
■
■
■
■
■
■
■
■
■
■
■
ed cells
ransfusion Support for Major Bleeding continued
Activity
Request suitable r
Consider the use of platelets
Consider the use of FFP
Table 6.2: T
Page 104
Transfusion Medicine Handbook 3rd Edition
,
illebrand factor
eby complementing
onectin ther
ecting multiple coagulation factor
elated mortality is high
Contains fibrinogen, FVIII, von W
FXIII, and fibr
FFP in corr
deficiencies
DIC-r
Comments
■
■
> 2.0
> 1.0 g/L
Maintain fibrinogen
Allow for 30 minutes thawing time
Dose: 1 unit per 30 kg body weight in adults (or
5 mL/kg paediatrics)
In obstetric bleeding maintain fibrinogen
g/L
Treat underlying cause (shock, hypothermia,
acidosis) if possible
Intervention
■
■
■
■
■
ransfusion Support for Major Bleeding continued
ecipitate
Activity
Consider the use of
cryopr
Suspect DIC
Table 6.2: T
Transfusion Medicine Handbook 3rd Edition
Page 105
6.2
Massive Transfusion Protocol (MTP)
The MTP is a multidisciplinary process by which blood components are obtained
rapidly for an exsanguinating patient. It is designed to provide clear guidance on the
management of massive blood loss and facilitate communication between the clinical
team and Blood Bank while streamlining the supply and administration of blood
components to a patient during what may be a stressful situation. The clinical team
is responsible for both activating the MTP and, when the crisis is over, inactivating
the MTP. Activation of the MTP is really only indicated where the patient is bleeding
so fast that goal-directed therapy is not practical.
The MTP instructs Blood Bank staff to prepare in advance a designated set or “box”
of blood components, provides confidence that those components will be available for
immediate release when required, and guides the clinical team in their administration.
After release of each box, Blood Bank will then prepare the next box but will not release
it until cal ed for. The MTP also recommends additional components if certain thresholds
are reached and the clinical team is responsible for requesting these.
Because hospitals serve unique patient populations and Blood Banks have differing
blood component stocks and ability to re-supply, the MTP for each DHB and affiliated
Blood Bank is specific, having been developed in consultation with local clinicians
responsible for managing these events. The principle however remains the same
and that is to provide the best possible transfusion support and ensure a common
understanding.
Protocol Activation Criteria
The massive transfusion protocol may be activated for a specific patient when the
following conditions are met:
■
There is massive bleeding with either shock or abnormal coagulation
■
The patient has been assessed as requiring the protocol by an experienced
clinician
In the setting of massive blood loss the following steps should be followed to activate
the massive transfusion protocol:
■
The patient is assessed by an experienced clinician
■
The patient is transfused 3 units of RBC or whole blood (either type-specific or
emergency O RhD negative)
■
The patient is reassessed and, if criteria are met, the MTP is activated
■
The clinician (or delegate) must notify Blood Bank of the activation, informing
them of the patient’s name, NHI and clinical area
■
The Blood Bank staff prepare and issue Box One and thereafter the MTP flow
chart is followed
■
Each MTP box will be made ready for issue upon release of the preceding box
but will only be issued upon request
Page 106
Transfusion Medicine Handbook 3rd Edition
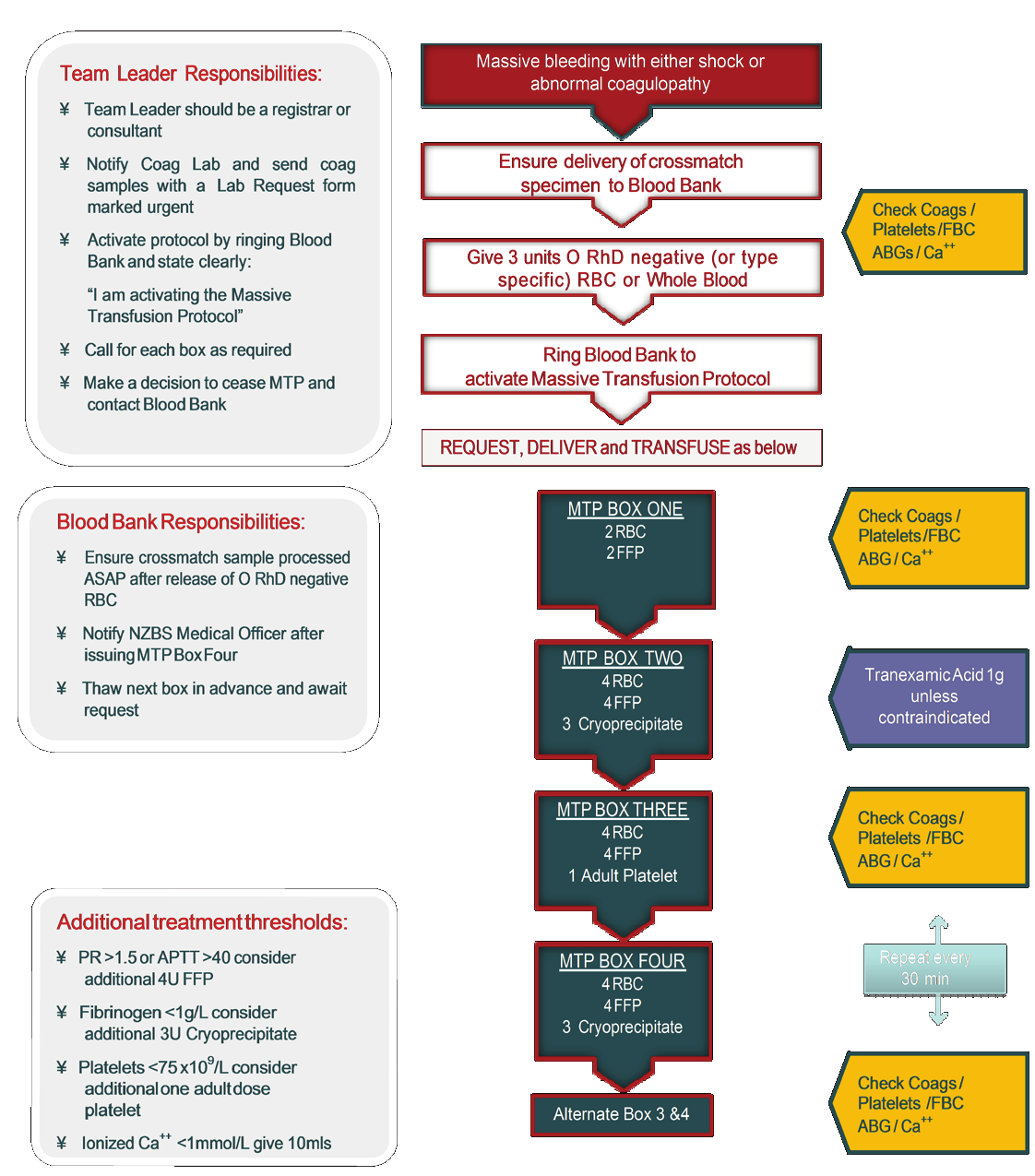
■
To avoid delay in receiving the blood components an orderly or “designated
runner” should be assigned
■
Alongside regular clinical assessment of the patient, a full blood count,
coagulation screen, ABG and serum calcium should be taken every 30 minutes
■
Based upon laboratory results consider the use of specific blood components
additional to those provided in standard MTP boxes
■
Blood Bank must be informed when the patient is moved to another clinical
department and/or the MTP is ceased.
Massive Transfusion Protocol Flowchart
Below is an example of a typical MTP flowchart. It is however important to be familiar
with the MTP used at your local DHB as there may be variations.
Transfusion Medicine Handbook 3rd Edition
Page 107
The Australian National Blood Authority
(NBA) Patient Blood Management Guidelines
recommend early activation of the MTP and that fibrinogen is maintained > 2.0 g/L in
obstetric patients. The role of permissive hypotension in these patients is uncertain as
this may compromise fetal well-being and, in the postpartum period, uterine contraction.
6.3
Complications of Acute Blood Loss Associated with Large Volume
Transfusions
When there is no pre-existing haemostatic problem, replacement of up to one blood
volume (8-10 units of blood in an adult) using red cells and non-plasma fluids is unlikely
to cause haemostatic problems due to dilution. Transfusion of much larger volumes
may however lead to:
■
Microvascular bleeding
When major blood loss and massive transfusion is complicated by microvascular
bleeding, with or without laboratory evidence of disseminated intravascular
coagulation (DIC), the platelet count should be maintained > 50 x 109/L. With
severe diffuse microvascular bleeding, a target platelet count ≥ 100 x 109/L is
recommended.
To avoid dilutional coagulopathy use of non-blood products should be restricted
until laboratory evidence that any haemostatic failure is corrected. Fresh frozen
plasma is indicated if the microvascular bleeding is accompanied by prolonged
PT/APTT > 1.5 times the mean control or the fibrinogen is < 1.0 g/L (< 1.5 g/L with
laboratory evidence of disseminated intravascular coagulation). With persisting
severe hypofibrinogenemia despite FFP, administration of cryoprecipitate is
recommended.
While there is no evidence that the prophylactic transfusion of fresh frozen plasma
or platelets to patients receiving large volume transfusions reduces the risk of
microvascular bleeding, these blood components are included emperically in
massive transfusion protocols in an attempt to maintain the platelet count > 50
x 109/L and PT/APTT < 1.5 times the mean control.
■
Hypocalcaemia
The citrate anticoagulant in some blood components (i.e., fresh frozen plasma)
binds ionised calcium. It should be noted that red cells in additive solution
contain only traces of citrate.
Usually the rapid metabolism of citrate by the liver prevents lowering of plasma
ionised calcium. In neonates and patients who are hypothermic, the combined
effects of hypocalcaemia and hyperkalaemia may be cardiotoxic. If there is ECG
or clinical evidence of hypocalcaemia, 5 mL of 10% calcium gluconate (for an
adult) should be given intravenously. If necessary the dose should be repeated
until the ECG is normal.
■
Hyperkalaemia
The plasma or additive solution in a unit of red cells or whole blood stored for
four to five weeks may contain 5-10 mmol of potassium. In the presence of
acidaemia and hypothermia this additional potassium load can lead to cardiac
arrest. Keeping the patient warm best prevents this problem.
■
Hypothermia
The rapid transfusion of blood at 4°C can lower the body’s core temperature by
several degrees. Keeping the patient warm is the best safeguard to prevent this
Page 108
Transfusion Medicine Handbook 3rd Edition
problem. A blood warmer should be used in adults receiving large volumes of
blood transfused at rates above 50 mL/kg/hour (in children above 15 mL/kg/hour).
It should be noted that hypothermic patients with a core body temperature < 35°C
may be functionally coagulopathic even though coagulation tests performed in
the laboratory at 37°C may be normal.
■
Acid-base disturbances
Despite the lactic acid content in transfused blood (1-2 mmol/unit), fluid
resuscitation usually improves acidosis in a shocked patient. In practice,
transfused citrate can contribute to metabolic alkalosis when large volumes of
plasma components are transfused.
■
Adult respiratory distress syndrome
The risk is minimised if tissue oxygenation is optimised by good perfusion and
over transfusion is avoided. The use of albumin solutions to maintain plasma
oncotic pressure is often stated to be important but controlled studies have not
proven any advantage of albumin solution over crystalloid fluids for resuscitation.
It should be noted that trauma (which may be the cause of major blood loss) is also
known to cause or contribute to hypothermia, acidosis and coagulopathy, and therefore
may lead to the problems described in association with massive transfusion.
6.4
Avoidable Haemostatic Problems in Elective Surgery
Any patient for whom elective surgery is planned must be asked about previous
episodes of abnormal bleeding. Underlying medical conditions, the taking of medication
that may be associated with impaired haemostatic function, or abnormal laboratory
haemostasis test results may require the postponement of elective surgery until the
abnormality has been identified or confirmed. Appropriate procedures including
consultation with specialists in haematology, anaesthesiology and cardiology should
be undertaken prior to surgery to minimise the perioperative risks for bleeding and, in
the case of anticoagulant or antiplatelet withdrawal, thromboembolism.
Congenital abnormalities of haemostasis such as haemophilia should be managed in
consultation with a specialist haemophilia centre.
6.4.1 Warfarin
Unless contraindicated, warfarin anticoagulation should be stopped early enough
before elective surgery to allow the prothrombin time (or INR; International Normalised
Ratio) to approach normal. This should be guided by a local protocol for preoperative
anticoagulant management, taking into account patient-related risk factors for both
thrombosis and bleeding. Bridging anticoagulation with either low molecular weight
heparin or heparin infusion may be indicated for patients with at least a moderate risk
of thrombosis. Prior to surgery where the INR is stable and therapeutic, withdrawal of
warfarin for 3 - 5 days is generally adequate to achieve reversal sufficient for surgery.
A longer period may be required in the presence of malnutrition or other factors
predisposing to vitamin K deficiency. The Australasian Society of Thrombosis and
Haemostasis (ASTH) has updated and published
Consensus Guidelines for Warfarin
Reversal in The Medical Journal of Australia 2013; 198 (4): 198-199. The
Guidelines
contain recommendations for the management of patients on long-term warfarin
undergoing invasive procedures. Additionally, the NZBS
Reversing Warfarin app
developed by Health Obs Ltd, available for android and iPhone, provides guidance
for managing patients undergoing elective and emergency surgery.
Transfusion Medicine Handbook 3rd Edition
Page 109
6.4.2 Non-Vitamin K-Dependent Oral Anticoagulants (NOAC)
The non-vitamin K-dependent oral anticoagulants, direct inhibitors of thrombin such
as dabigatran (Pradaxa) or factor Xa such as rivaroxaban (Xarelto) and apixiban
(Eliquis), should be stopped early enough before elective surgery to allow reversal of
the anticoagulant effect. This should be guided by a local protocol for preoperative
anticoagulant management with, in the case of dabigatran, particular attention paid
to the patient’s renal function and hence elimination of the drug. The Australasian
Society of Thrombosis and Haemostasis has published
New Oral Anticoagulants:
A Practical Guide on Prescription, Laboratory Testing and Peri-procedural/Bleeding
Management in The Internal Medicine Journal 2014; 44: 525-536. The
Guide contains
recommendations for the management of patients taking NOAC undergoing invasive
procedures. Similarly, the Pharmaceutical Management Agency of the New Zealand
Government (PHARMAC), has produced a concise document,
Guidelines for Testing
and Perioperative Management of Dabigatran (www.pharmac.govt.nz). Additionally,
the
Rivaroxaban app and the
Dabigatran app developed by Health Obs Ltd, available
for android and iPhone, provide guidance for managing patients undergoing elective
and emergency surgery.
Table 6.3: Pharmacologic Properties of Non-Vitamin K-Dependent Oral Anticoagulants
Dabigatran
Rivaroxaban
Apixaban
Peak level
2 hours
1.5 - 4 hours
3 - 4 hours
Renal clearance
80%
66%
27%
11 - 17 hours
5 - 9 hours
8 - 15 hours
Half-life
(longer in elderly and, especially dabigatran, with impaired renal function)
Table 6.4: Suggested Approach for the Withdrawal of Non-Vitamin K-Dependent Oral
Anticoagulants Prior to Elective Surgery
Dabigatran
Rivaroxaban
Apixaban
Normal or mild renal
impairment1
2 days
2 days
2 days
(eGFR > 50 mL/min)
Moderate renal
impairment1
4 days
3 days
3 days
(eGFR 30 - 50 mL/min)
Severe renal impairment2
5 days
4 days
3 days
(eGFR < 30 mL/min)
1 For surgery with minimal bleeding risk, NOAC withdrawal may be shortened by 1 day. For surgery with
high bleeding risk, NOAC withdrawal should be prolonged by 1 day.
2 NOAC use is contraindicated or generally not recommended in the setting of severe renal impairment and
should not be restarted post-operatively.
Page 110
Transfusion Medicine Handbook 3rd Edition
6.4.3 Aspirin
The antiplatelet effect of aspirin is mediated via irreversible acetylation of platelet
cyclo-oxygenase, specifically COX-1, and the resulting inhibition of thromboxane A2
synthesis. Aspirin is indicated for the management of ischaemic atherosclerotic vascular
disease including coronary artery and cerebrovascular disease and is used primarily
in secondary prevention. Aspirin is used in combination with ADP-receptor inhibitors
for the management of acute coronary syndromes (ACS) and for thromboprophylaxis
following stent implantation. It may also be used for the management of some
rheumatological disorders. While a single 75 mg dose of aspirin may impair platelet
function for several days, the effect is less than with newer antiplatelet agents and
most surgery can be performed after discontinuing aspirin at most the day prior to
surgery. Prior to neurosurgery, transurethral prostatectomy or other invasive procedures
with a major risk of bleeding or complications from bleeding, aspirin should (unless
contraindicated) be stopped 3 days before planned surgery. If aspirin-induced platelet
defect contributes to abnormal bleeding, desmopressin and tranexamic acid are likely
to be effective in controlling haemostasis. Refer to Section 8.6:
Desmopressin and
Section 8.7:
Tranexamic Acid for further information on these agents including dosing
guidelines. Platelet transfusion is seldom necessary.
6.4.4 Non-steroidal Anti-inflammatory Drugs (NSAID)
Non-steroidal anti-inflammatory drugs cause reversible, but non-selective, inhibition
of cyclo-oxygenase, both COX-1 and COX-2. The effects usually last for hours,
as opposed to lasting days with aspirin. Normal platelet function is usually rapidly
restored once the NSAID is stopped. The length of time that these medications
should be stopped prior to an invasive procedure varies as the antiplatelet effect is
dependent on the half-life of the NSAID being used. If NSAID-induced platelet defect
contributes to abnormal bleeding, desmopressin and tranexamic acid may be effective
in controlling haemostasis. Platelet transfusion is seldom necessary. The effect of both
desmopression and platelet transfusion will be reduced in the presence of active drug.
6.4.5 P2Y Adenosine Diphosphate (ADP) Receptor Inhibitors
12
Clopidogrel and the other thienopyridines, ticlopidine (not commonly in use) and
prasugrel (Effient), irreversibly inhibit the P2Y subtype of ADP receptor on platelet
12
cell membranes. Ticagrelor (Brilinta), a cyclopentyltriazolopyrimidine reversibly inhibits
the P2Y ADP receptor. Clopidogrel is indicated for the management of ischaemic
12
atherosclerotic vascular disease including coronary artery and cerebrovascular disease.
The newer agents prasugrel and ticagrelor are indicated for the management of acute
coronary syndromes (ACS) and as prophylaxis against stent thrombosis, usually in
combination with aspirin. In some situations triple therapy in combination with an
anticoagulant is indicated. P2Y ADP receptor inhibitors should be used with caution
12
in patients at increased risk of bleeding such as in the setting of trauma, surgery or
other pathological conditions of haemostasis. Unless contraindicated, clopidogrel
should be discontinued at least 5 days prior to surgery where an antiplatelet effect
is undesirable. Prasugrel should be discontinued 7 days prior to surgery. Ticagrelor,
although reversible, has a pronounced effect on platelet function and should be
discontinued at least 3 days prior to semi-urgent surgery and 5 days prior to elective
surgery. If P2Y ADP receptor-induced platelet defect contributes to abnormal
12
Transfusion Medicine Handbook 3rd Edition
Page 111
bleeding, tranexamic acid together with a 1 - 2 unit platelet transfusion is likely to be
effective in controlling haemostasis. Ticagrelor has a long half-life and inhibition of
even transfused platelets may occur. An effect on transfused platelets may be seen
for up to 6 hours following a loading dose of prasugrel. In patients with ACS who
have recently received a coronary stent, platelet transfusion carries a risk of arterial
and stent thrombosis and should therefore be restricted to situations involving serious
bleeding or high bleeding risk.
6.4.6 Platelet Glycoprotein IIb (GPIIb) and IIIa (GPIIIa) Inhibitors
The platelet glycoproteins GPIIb and GPIIIa have important roles in normal haemostasis
and pathological thrombosis. The peptides tirofiban (Aggrastat) and eptifibatide
(Integrilin) and the monoclonal antibody abciximab (ReoPro) inhibit GPIIb and GPIIIa
receptors. Inhibition of platelet aggregation occurs through blocking the final common
pathway, the cross-bridging of platelets, following binding of fibrinogen to the activated
GPIIb/IIIa receptor. These agents are indicated, in combination with aspirin and heparin
anticoagulation, for the management of acute coronary syndromes (ACS) managed
medically with or without percutaneous coronary intervention (PCI). Current applicability
is however limited following the introduction of routine dual antiplatelet therapy using
P2Y12 ADP receptor blockers and percutaneous coronary stenting. Tirofiban and
eptifibatide bind reversibly to GPIIb/IIIa receptors and the antiplatelet effect, in the
absence of moderate renal impairment, recovers within 4 - 8 hours following cessation
of treatment. In contrast, platelet binding by abciximab is irreversible and recovery of
platelet function is delayed for 24 - 48 hours. By reducing thrombin burst generation,
abciximab also inhibits normal coagulation and major bleeding may be observed
within 12 hours of therapy, particularly following cardiac surgery. The antiplatelet
effects of abciximab can be reversed by platelet transfusion. A small percentage of
patients (≤ 1%) may experience acute severe thrombocytopenia following treatment
with GPIIb/GPIIIa inhibitors.
Table 6.5: Pharmacologic Properties of Antiplatelet Agents
Aspirin
Clopidogrel
Prasugrel
Ticagrelor
Mode of action
Prodrug
Prodrug
Prodrug
Direct acting
Active drug t1/2
2 - 4 hours
30 mins
7 hours1
7 - 9 hours
Prodrug t1/2
NA2
6-8 hours
NA2
NA
Inhibition type
Irreversible3
Irreversible3
Irreversible3
Reversible4
Grade of effect
Mild
Moderate
Strong
Strong
Specific antidote
No
No
No
No
1 Range 2 - 15 hours.
2 Aspirin and prasugrel are rapidly converted to an active metabolite.
3 The duration of inhibition is for the life span of the plalelet, i.e., 7-10 days. However, the effect on platelet-
mediated haemostatic function declines more rapidly as approximately 10-15% of the circulating platelet
pool is replaced daily.
4 The duration of inhibition is dependent on the half-life (t ). In general, it can be expected that after two
1/2
half-lives the effect of ticagrelor has reduced to 25%. After four to five half-lives the remaining effect of
ticagrelor is <
5%.
Page 112
Transfusion Medicine Handbook 3rd Edition
Table 6.6: Managing Bleeding in Patients on Antiplatelet Agents
Aspirin
Clopidogrel
Prasugrel
Ticagrelor
Withdrawal
pre-surgery
0 - 1 day1
5 days
7 days
5 days
Bleeding reversal2
Tranexamic acid
Yes
Yes
Yes
Yes
Desmopressin
Yes
Possible effect
No effect
No effect
Platelets3
Seldom required
1 unit
2 units
2 units
1 Prior to neurosurgery, transurethral prostatectomy or other invasive procedures with a major risk of
bleeding or complications from bleeding, aspirin should (unless contraindicated) be stopped for 3 days.
2 Where appropriate, local haemostatic measures should be used including mechanical compression,
topical application of tranexamic acid and surgical/radiological intervention to identify sources of bleeding.
3 Repeated doses may be required to achieve or maintain effect.
6.5
Oral Anticoagulant Induced Bleeding or Overdose
6.5.1 Warfarin
Withholding warfarin along with the judicious use of oral vitamin K1 are the management
options of choice unless rapid reversal of anticoagulation is required. Although
intravenous administration of vitamin K1 produces a more rapid response (onset
within 6 - 8 hours), at 24 hours both routes achieve a similar correction of INR. The
intravenous route may rarely be associated with anaphylaxis. The full effect of vitamin
K1 in reducing the INR takes up to 24 hours even when given in large doses. For
immediate reversal of clinically significant bleeding, treatment with Prothrombinex®-VF
covers the period until vitamin K1 achieves full effect. Subsequent doses of vitamin
K1 may be necessary for maintaining correction of the INR achieved by coagulation
factor replacement. Prothrombinex®-VF is a human prothrombin complex concentrate
(PCC) containing factors II, IX and X together with only low levels of factor VII. The
use of fresh frozen plasma should therefore be considered as a source of factor VII in
life-threatening or critical organ bleeding. If no PCC is available, fresh frozen plasma
should be transfused however this is less effective. The FFP dose for an adult in this
setting is 15 mL/kg.
The ASTH has updated and published
Consensus Guidelines for Warfarin Reversal in The Medical Journal of Australia, Med J Aust 2013; 198 (4): 198-199, from which
the following table is adapted.
Transfusion Medicine Handbook 3rd Edition
Page 113
Table 6.7: Managing Overdose or Bleeding in Patients on Warfarin Therapy
Clinical setting
Action
INR greater than the
■
Reduce or omit next dose of warfarin and resume
therapeutic range
therapy at a lower dose when INR approaches
but less than 4.5; no
therapeutic range
bleeding
■
If INR is only minimally above the therapeutic range (up
to 10%) dose reduction may not be necessary
INR greater than 4.5
■
Stop warfarin and consider reasons for elevated INR
but less than 10.0; no
■
If bleeding risk is high1, give vitamin K1 1.0-2.0 mg
bleeding
orally or 0.5-1.0 mg intravenously
■
Measure the INR within 24 hours (vitamin K1 effect on
INR expected within 6-12 hours) and monitor closely
for 1 week
■
Restart warfarin at a reduced dose once the INR
approaches therapeutic range
INR greater than 10.0;
■
Stop warfarin
no bleeding2
■
Give 3.0-5.0 mg vitamin K1 orally or intravenously
■
If bleeding risk is high1, consider Prothrombinex®-VF
15-30 IU/kg
■
Measure the INR within 24 hours (vitamin K1 effect on
INR expected within 6-12 hours) and monitor closely
for 1 week
■
Restart warfarin at a reduced dose once the INR
approaches therapeutic range
Life-threatening3
or
■
Stop warfarin
critical organ bleeding ■ Give 5.0-10.0 mg IV vitamin K1,
and Prothrombinex®-
with an INR ≥ 1.5
VF 50 IU/kg4
and fresh frozen plasma 150-300 mL
■
If Prothrombinex®-VF is unavailable, administer fresh
frozen plasma 15 mL/kg
■
Assess patient continuously until INR is reversed and
bleeding stops
Clinically significant
■
Stop warfarin
(non-life-threatening)
■
Give 5.0-10.0 mg IV vitamin K1,
and Prothrombinex®-
bleeding with an INR
VF 35-50 IU/kg
≥ 2.0
■
If Prothrombinex®-VF is unavailable, administer fresh
frozen plasma 15 mL/kg
■
Assess patient continuously until INR is reversed and
bleeding stops
Minor bleeding with
■
Omit warfarin and repeat INR the following day
any INR < 10.0
■
Adjust warfarin dose to maintain INR in therapeutic
range
■
If bleeding risk is high1 or INR > 4.5, consider vitamin
K1 1.0-2.0 mg orally or 0.5-1.0 mg intravenously
Page 114
Transfusion Medicine Handbook 3rd Edition
1 Risk factors for major bleeding include recent major bleed within previous four weeks, major surgery within
previous two weeks, platelet count <
50 x 109/L, known liver disease, concurrent antiplatelet therapy.
2 New Zealand laboratories generally report INR values up to 8.0. Above this, results are reported as INR >
8.0.
3 Includes intracranial bleeding.
4 Consider a Prothrombinex®-VF dose less than 50 IU/kg when INR is 1.5-1.9.
The NZBS
Reversing Warfarin app developed by Health Obs Ltd, available for android
and iPhone, also provides guidance for managing patients with bleeding.
6.5.2 Non-Vitamin K-Dependent Oral Anticoagulants (NOAC)
There is limited clinical data on reversal of the direct thrombin inhibitor dabigatran
and the factor Xa inhibitors rivaroxaban and apixaban. As there is no specific reversal
agent available, the mainstay of current management for drug-related bleeding is
withholding NOAC together with best supportive care for local control of bleeding and
maintenance of haemodynamic stability. The anticoagulant effect will not be reversed
by the administration of vitamin K or infusion of plasma. Idarucizumab, a specific
intravenous reversal agent for dabigatran, is showing promising early results in clinical
trials. The antibody fragment completely reverses within minutes the anticoagulant
effect of dabigatran.
The ASTH has published
New Oral Anticoagulants: A
Practical Guide on Prescription,
Laboratory Testing and Peri-procedural/Bleeding Management in The Internal Medicine
Journal 2014; 44: 525-536. The
Guide contains recommendations for the management
of patients taking NOAC with bleeding complications. Similarly, PHARMAC, has
produced a concise document,
Guidelines for Management of Bleeding with
Dabigatran (www.pharmac.govt.nz). The recommendations may change as new
evidence becomes available. Additionally, the
Rivaroxaban app and the
Dabigatran app developed by Health Obs Ltd, available for android and iPhone, provide guidance
for managing patients with bleeding.
Table 6.8: Managing Bleeding in Patients on Non-Vitamin K-Dependent Oral Anticoagulant
Therapy
Clinical setting
Action
General measures for
■
Initiate standard resuscitation procedures as required
any patient bleeding
■
Take samples for FBC, creatinine, group and screen,
on NOAC therapy
routine coagulation studies
■
Dabigatran - APTT, thrombin time; consider dabigitran
level using a dilute thrombin clotting time (Hemoclot)
assay
■
Rivaroxaban - PT1, drug specific anti-Xa level
■
Apixaban - drug specific anti-Xa level (if available)
Minor bleeding
■
Local haemostatic measures
- mechanical compression
- topical tranexamic acid
■
Delay next dose of NOAC
■
Consider appropriateness of continuing therapy in
consultation with prescribing physician
Transfusion Medicine Handbook 3rd Edition
Page 115
Table 6.8: Managing Bleeding in Patients on Non-Vitamin K-Dependent Oral Anticoagulant
Therapy continued
Clinical setting
Action
Clinically significant2
■
Stop NOAC
(non-life-threatening)
■
Administer oral charcoal if NOAC ingestion < 2 h prior
bleeding
■
Local haemostatic measures
- mechanical compression
- surgical/radiological intervention to identify and
- treat source of bleeding
■
Maintain hydration to aid drug clearance
■
Transfusion support
- RBC as indicated by haemoglobin
- PLT if < 50 x 109/L or antiplatelet therapy
- FFP only if concerned about dilutional coagulopathy
■
Consider use of tranexamic acid 15-30 mg/kg IV +/-
infusion for mucosal bleeding
Life-threatening
■
Stop NOAC
or critical organ
■
Administer oral charcoal if NOAC ingestion < 2 h prior
bleeding3
■
Local haemostatic measures
- mechanical compression
- surgical/radiological intervention to identify and
treat source of bleeding
■
Maintain hydration to aid drug clearance
■
Transfusion support
- RBC as indicated by haemoglobin
- PLT if < 100 x 109/L or antiplatelet therapy
- FFP only if concerned about dilutional
coagulopathy
■
Consider use of the following pro-haemostatic agents4
- Tranexamic acid 15-30 mg/kg IV +/- infusion for
mucosal bleeding
- Prothrombinex®-VF 25-50 IU/kg
- FEIBA 50 IU/kg
■
Consider dialysis for dabigatran-related bleeding5
■
Assess patient continuously until bleeding stops
1 Using a thromboplastin sensitive to rivaroxaban.
2 Clinically significant bleeding - reduction in Hb ≥
20 g/L, transfusion of ≥
2 units of RBC.
3 Life-threatening bleeding with hypotension not responding to resuscitation or bleeding in critical area or
organ (intraocular, intracranial, intraspinal, compartment syndrome, retroperitoneal or pericardial).
4 This is an off-license use of Prothrombinex®-VF and FEIBA and the risk of thrombotic complications with
these agents when used for this indication is unclear. Their use is supported by laboratory data and clinical
evidence in animals but clinical evidence in humans supporting an improvement in clinical outcomes is lacking.
5 Dialysis is indicated if dabigatran level is high as indicated by excessively prolonged APTT >
80 sec or
dabigatran level >
500 ng/mL and/or impaired renal function. Four hours of dialysis will reduce drug level
by approximately 60%.
Page 116
Transfusion Medicine Handbook 3rd Edition
6.6
Thrombolytic Therapy
Although bleeding is not a common complication of fibrinolytic therapy at normal
doses, the risk is not predicted by laboratory monitoring. If there is serious bleeding,
fresh frozen plasma and cryoprecipitate will raise a low fibrinogen level.
An anti-fibrinolytic agent such as tranexamic acid should only be used if life-threatening
bleeding is encountered. As large clots may form at the site of bleeding, these agents
are contraindicated in renal tract bleeding. It should however be noted that the use of
anti-fibrinolytic agents is not contraindicated in the presence of intracranial bleeding
following thrombolysis.
6.7
Disseminated Intravascular Coagulation (DIC)
In this syndrome there is generation of thrombin leading to consumption of circulating
coagulation factors and platelets with subsequent fibrin deposition. Ischaemic
organ damage particularly in the renal circulation can occur due to microthrombi.
The treatment of DIC involves supportive care while treating the underlying primary
condition. In the presence of active bleeding, or where there is a high risk of major
bleeding such as prior to some invasive procedures, transfusion support to replace
coagulation factors and platelets is likely to be appropriate. If the patient is bleeding
and there are no problems with volume replacement, fresh frozen plasma is optimal
as it contains a full spectrum of coagulation factors. Where real concern exists
for intravascular fluid overload, Prothrombinex®-VF may be considered, as the
reconstituted volume for infusion is smaller. Correction of the coagulation defect is
however only likely to be partial and Prothrombinex®-VF is generally contraindicated in
the presence of DIC. For bleeding patients with severe hypofibrinogenaemia despite
FFP replacement, cryoprecipitate should be infused keeping the fibrinogen level > 1.5
g/L. Antithrombin concentrates may be considered where DIC is secondary to sepsis
or in cases of thrombosis-predominant DIC treated with therapeutic dose heparin. A
platelet count of 10 - 20 x 109/L can usually be tolerated in the absence of bleeding
while transfusion is recommended with a platelet count < 50 x 109/L in the presence
of active bleeding or prior to invasive procedures. Where the procedure is neuraxial a
higher platelet is likely to be desirable.
The management of DIC requires careful coordination between the treating clinician
and NZBS to ensure adequate supplies of blood components and plasma products
are available and that these are used based upon relevant coagulation tests.
6.8
Cardiopulmonary Bypass
Cardiopulmonary bypass usually impairs haemostatic mechanisms and, as a result,
bleeding complications may be severe. The effect is especially pronounced for patients
who have been on prolonged bypass for more than 2 hours, in ‘redo’ operations or
with surgery for infective endocarditis. A number of mechanisms are involved including
haemodilution, effects on platelet function, reduced levels of coagulation factors,
intraoperative heparin anticoagulation, induced hypothermia, acidosis, hyperfibrinolysis
and preoperative anticoagulation or antiplatelet therapy.
Routine laboratory tests of coagulation do not accurately predict the full nature and
clinical importance of the haemostatic defect. Intra-operative point-of-care testing with
an activated clotting time (ACT) assay, together with viscoelastic global assessments
Transfusion Medicine Handbook 3rd Edition
Page 117
of coagulation using whole blood thromboelastography (TEG) or thromboelastometry
(TEM), is increasingly being used alongside PT/APTT and fibrinogen results.
Platelet transfusion is indicated if, despite heparin reversal with protamine sulphate,
there is diffuse microvascular bleeding with platelets < 100 x 109/L or suspected
platelet dysfunction. In the presence of non-surgical bleeding, fresh frozen plasma and
cryoprecipitate may help to correct prolonged clotting times, improve haemostasis and
reduce transfusion requirements. The routine use of fresh frozen plasma, cryoprecipitate
or platelets at the end of bypass does however not reduce transfusion requirements.
Aprotinin (Trasylol®) is no longer available due to concerns regarding effect on renal
function. Tranexamic acid is commonly used as an antifibrinolytic and has been shown
to reduce red cell transfusion requirement.
Red cell components for patients undergoing cardiac bypass surgery Literature review provides no evidence supporting the use of fresh blood for cardiac
bypass surgery. Accordingly, NZBS policy on the provision of red cell components,
including whole blood, for use in recipients undergoing cardiopulmonary bypass
surgery is as follows:
■
Adult Cardiac Bypass Surgery
Resuspended red cells less than 14 days old will normally be made available for
adult patients as a replacement for blood loss. Resuspended red cells less than
10 days old will normally be made available for patients with renal impairment.
It is the responsibility of the treating clinician to indicate this requirement to the
Blood Bank.
■
Paediatric Cardiac Bypass Surgery
Resuspended red cells less than 5 days old will normally be made available for
paediatric patients as a replacement for blood loss. Where possible, whole blood
less than 2 days old will be provided for the purpose of bypass circuit priming.
6.9
Haemolytic Disease of the Fetus and Newborn (HDFN)
HDFN occurs when maternal IgG antibodies (most commonly anti-D) travel across the
placenta and bind to fetal red cells having the corresponding antigen. The affected fetal
red cells may be destroyed by the fetal reticuloendothelial system causing extravascular
haemolysis and fetal anaemia.
The antibodies that cause HDFN are produced either as a consequence of earlier
pregnancies (when fetal red cells with paternally derived antigens that the mother lacks,
enter the maternal circulation during pregnancy) or because of a previous transfusion.
In the most severe cases of HDFN the fetus may die in utero or be born with severe
anaemia that requires exchange transfusion. There may also be severe neurological
damage (kernicterus) as a result of a high bilirubin level.
Anti-D, an antibody to the RhD antigen, is the most important cause of HDFN. Clinically
significant IgG antibodies against other blood group antigens can also be responsible
for causing HDFN for example anti-c, -K, -E, -Fya. These antibodies occur in about
0.5% of pregnancies and may occasionally cause severe haemolysis. Although ABO
incompatibility between mother and fetus is common, severe HDFN due to IgG anti-A
and anti-B antibodies is very rare in New Zealand.
Page 118
Transfusion Medicine Handbook 3rd Edition
10
Screening in Pregnancy
All pregnant women should have the following antenatal testing performed:
■
ABO and RhD group following booking for antenatal care at 12 - 16 weeks.
This identifies RhD negative mothers.
■
Red cell antibody screen to test for antibodies that may cause HDFN. If
an antibody is detected at booking, it should be monitored throughout the
pregnancy in case the level of antibody increases.
■
If a significant maternal antibody is found it may be useful to test the father’s
red cells to see if they carry the antigen against which the antibody is directed.
If homozygous for the antigen concerned, the fetus will also be positive. If
heterozygous, there is a 50% chance that the fetus will be positive.
Further information can be found in the ANZSBT/RANZCOG
Guidelines for Blood
Grouping & Antibody Screening in the Antenatal and Perinatal Setting (2007) upon
which the following table is based.
Table 6.9: Timetable for Routine Antenatal Blood Group and Antibody Screen
Testing Requirements
Testing Interval / Comments
Blood Group - ABO and RhD
All pregnancies
■
Initial visit and for pretransfusion testing
Antibody Screening
All pregnancies
■
Initial visit and for pretransfusion testing
RhD negative females
■
For those RhD negative females who will receive
anti-D immunoglobulin (at 28 weeks or at the
time of any sensitising event), the blood sample
must be collected prior to its administration
Antibody Identification
All pregnancies
■
At initial antibody detection
Antibody Titration / Quantitation
Rh antibodies and other
■
In the event that clinically significant antibodies
potentially clinically significant
are detected, further testing is indicated at
antibodies capable of causing
intervals no greater than 4 weeks
HDFN
■
Seek specialist advice
Management of HDFN
It is important that women with potentially severe HDFN are referred without delay
to a specialist obstetric unit. The referral should be made prior to 20 weeks in those
patients who have had a previously affected baby. Management of an affected fetus
may include intrauterine transfusion, early delivery, phototherapy and exchange
transfusion.
Transfusion Medicine Handbook 3rd Edition
Page 119
Prevention of HDFN Due to Anti-D
The minimum volume of RhD positive red cells that will immunise a RhD negative
woman is of the order of 0.1 - 0.25 mL. Clinical studies indicate that the administration
of anti-D immunoglobulin to a RhD negative mother within 72 hours of the birth of a
RhD positive infant reduces the incidence of Rh isoimmunisation from 12 - 13% to
1 - 2%. A small number (1.5 - 1.8%) of RhD negative mothers are immunised by their
RhD positive fetus despite postpartum administration of anti-D immunoglobulin. Studies
have also shown that this number can be reduced to < 1.0% by routine antenatal
prophylaxis with anti-D immunoglobulin at 28 and 34 weeks of pregnancy. Although
definitive studies have not been performed, guidelines promote the use of anti-D
immunoglobulin for other potential y sensitising obstetric events in which fetomaternal
haemorrhage is known to occur.
Anti-D immunoglobulin should not be given to RhD negative women with detectable
anti-D except where the antibody is passively acquired due to prior antenatal
administration. If unsure whether the anti-D detected in the mother’s blood is passively
acquired or preformed, the treating clinician and/or a NZBS Transfusion Medicine
Specialist should be consulted. If there is continuing doubt, anti-D immunoglobulin
should be administered. Although there is no benefit in administering anti-D
immunoglobulin to a woman who is already sensitised to RhD antigen, there is no
more risk than when it is given to a woman who is not sensitised.
The following table is based on the 2003 Australian NBA
Guidelines on the Prophylactic
Use of RhD Immunoglobulin (Anti-D) in Obstetrics and the 2013 BCSH
Guideline for the
Use of Anti-D Immunoglobulin for the Prevention of HDFN. In 2011 the Royal Australian
and New Zealand College of Obstetricians and Gynaecologists (RANZCOG) endorsed
the 2003 NBA Guidelines, however it should be noted that the NZ Ministry of Health
does not recommend routine antenatal anti-D prophylaxis (RAADP).
Table 6.10: Indications for Use of Anti-D Immunoglobulin for the Prevention of HDFN
(unless the fetus is confirmed to be RhD negative)
Timing
Clinical Indication
Routine antenatal
A RhD negative woman, not previously immunised
prophylaxis1
to produce anti-D; one dose at each of 28 and 34
weeks gestation.2
Anti-D immunoglobulin dose: 625 IU (125 µg)
Potentially sensitising event
A RhD negative woman, not previously immunised
during first trimester up
to produce anti-D, with an obstetric indication.
to and including 12 weeks
■
Uterine bleeding where this is repeated, heavy
gestation
or associated with abdominal pain3
■
Miscarriage3
■
Termination of pregnancy
■
Ectopic pregnancy
■
Molar pregnancy
Anti-D immunoglobulin dose: 250 IU (50 µg)4
Page 120
Transfusion Medicine Handbook 3rd Edition
Table 6.10: Indications for Use of Anti-D Immunoglobulin for the Prevention of HDFN
(unless the fetus is confirmed to be RhD negative) continued
Timing
Clinical Indication
Potentially sensitising event
A RhD negative woman, not previously immunised
beyond first trimester5
to produce anti-D, with an obstetric indication.
■
Amniocentesis, chorionic villus sampling, and
intrauterine fetal blood sampling
■
Antepartum haemorrhage (or unexplained
uterine pain)
■
External cephalic version (performed or
attempted)
■
Abdominal trauma sufficient to cause FMH
■
Ectopic pregnancy
■
Molar pregnancy
■
Intrauterine death or stillbirth
■
Miscarriage, threatened miscarriage
■
Termination of pregnancy
Anti-D immunoglobulin dose: 625 IU (125 µg)
Postpartum prophylaxis6
A RhD negative woman, not previously immunised
to produce anti-D, who gives birth to a RhD positive
baby.
Anti-D immunoglobulin dose: 625 IU (125 µg) with
additional dose(s) indicated where fetomaternal
haemorrhage is > 6 mL fetal red cells
1 Routine antenatal anti-D prophylaxis (RAADP) should be administered regardless of, and in addition to,
prophylaxis given for a potentially sensitising event.
2 A sample for antibody testing should be taken prior to administration of anti-D immunoglobulin at 28 weeks.
3 Before 12 weeks gestation, in cases of either spontaneous complete miscarriage where the uterus is
not instrumented or mild painless vaginal bleeding, the risk of fetomaternal haemorrhage is negligible.
4 For multiple pregnancies the recommended anti-D immunoglobulin dose is 625 IU (125 µg).
5 Routine antenatal prophylaxis does not preclude prophylaxis for a potentially sensitising event.
6 Routine antenatal prophylaxis or prophylaxis for a potentially sensitising event does not preclude postpartum
prophylaxis.
Laboratory Assessment of Fetomaternal Haemorrhage (FMH)
■
The Kleihauer acid elution test is widely used to test for FMH and can detect
bleeds of 0.1 mL or less. False results can occur with the test and it is less
reliable in the first two trimesters of pregnancy when there is an increase in the
level of fetal haemoglobin in the maternal red blood cells. Clinicians using this
test should be aware of these limitations. Anti-D immunoglobulin should still be
administered if a negative Kleihauer test is obtained.
■
There is no need to assess the size of FMH in pregnancies of 20 weeks gestation
or less, as the standard 250 IU (up to and including 12 weeks) and 625 IU
(beyond 12 weeks, up to and including 20 weeks) dose of anti-D immunoglobulin
will sufficiently cover the maximum likely FMH for the gestation.
■
It is appropriate to assess the size of FMH after 20 weeks gestation. A blood
sample should be taken from the mother as soon as possible after the potentially
sensitising event and before the dose of anti-D immunoglobulin is given.
Transfusion Medicine Handbook 3rd Edition
Page 121
■
Results reporting the fetal bleed in mL of red cells should be promptly available
so that anti-D immunoglobulin can be given within 72 hours of the FMH.
■
For large bleeds greater than 4 mL, repeat testing should be performed at 48
hours following each intravenous dose of anti-D or 72 hours following each
intramuscular dose of anti-D immunoglobulin to check for clearance of fetal
red cells and, if still positive, additional dose(s) given.
■
Flow cytometry, a more accurate and reproducible method for measuring FMH,
is not yet widely available.
Timing, Dose and Route of Anti-D Immunoglobulin
■
Anti-D immunoglobulin prophylaxis should be given as soon as possible and
always within 72 hours of a potentially sensitising event.
■
If anti-D immunoglobulin has not been offered within 72 hours, a dose given
within 10 days may provide some protection.
■
A 625 IU dose of anti-D immunoglobulin will protect against a FMH of up to
6 mL of RhD positive red cells (equivalent to 12 mL of blood). Where testing
shows that a FMH of greater than 6 mL has occurred, additional dose(s) of
anti-D immunoglobulin must be administered. In this situation, the recommended
minimum dose is 100 IU per mL RhD positive red cells.
■
Where more than 1250 IU of anti-D immunoglobulin (two 625 IU vials of Rh(D)
Immunoglobulin-VF) is indicated, consultation with a NZBS Transfusion Medicine
Specialist is recommended.
■
For bleeds requiring a large anti-D immunoglobulin dose, the maximum
recommended administration rate is 3000 IU every 8 hours to reduce the risk of
an adverse reaction arising from rapid clearance of fetal RhD positive red cells.
As it is recommended that no more than 4 mL is injected intramuscularly at each
site, Rhophylac®, an anti-D immunoglobulin product suitable for intravenous
administration, may be an appropriate alternative.
■
Recipients who have a moderate or severe thrombocytopenia should not
receive intramuscular injections. The standard anti-D immunoglobulin provided
by NZBS, Rh(D) Immunoglobulin-VF, is suitable for intramuscular use only. It
may be given by intramuscular or subcutaneous route; it must not be given
intravenously. Rhophylac, an anti-D immunoglobulin product suitable for
intravenous administration is held at NZBS sites and can be obtained following
consultation with a NZBS Transfusion Medicine Specialist/Medical Officer.
■
There is some evidence to suggest that intramuscular administration of anti-D
immunoglobulin may be associated with an increased risk of lack of effect in
patients with a body mass index (BMI) > 30. An Expert Panel Consensus Position
Statement (available online at www.transfusion.com.au/node/612) provides
recommendations regarding the use of anti-D immunoglobulin in these patients.
The following is recommended where a woman has received an appropriate dose of
anti-D immunoglobulin followed by a further risk event for immunisation.
■
Where the previous dose was given 2 or more weeks before, a further dose(s)
of anti-D immunoglobulin should be offered.
Page 122
Transfusion Medicine Handbook 3rd Edition
■
Where the previous dose was given less than 2 weeks before, a further dose of
anti-D immunoglobulin should be offered where the pregnancy is more than 20
weeks gestation. Where testing indicates a FMH greater than 6 mL, additional
dose(s) of anti-D immunoglobulin must be administered.
■
Where there is continual uterine bleeding that is judged clinically to represent the
same risk event, further dose(s) of anti-D immunoglobulin should be offered at a
minimum of 6 weekly intervals. In pregnancies > 20 weeks gestation, estimation
of FMH should be undertaken at 2 weekly intervals and the presence of fetal
cells should prompt an additional anti-D immunoglobulin dose to cover the
volume of FMH.
Further observations and comment on the guidelines for the use of anti-D
immunoglobulin are available in the NZBS Clinical Compendium.
Non-invasive Fetal Genotyping
A maternal blood sample may be used for non-invasive testing of cell-free fetal DNA
to predict the fetal red cell phenotype. This has particular value in identifying a fetus
that is unlikely to be at risk of HDFN. In the case of a sensitised RhD negative woman,
the absence of the RhD gene predicts for a RhD negative fetus. As such, aggressive
monitoring of the mother’s anti-D titre and the health of the fetus is unnecessary. Fetal
RhD status is also useful in determining whether anti-D immunoglobulin prophylaxis
is required. Rarely, such as when genetic changes lead to gene silencing rather than
deletion, genotype determination does not correlate with the antigen expression on
the fetal red cells.
Certain criteria need to be met before obtaining fetal DNA for genotyping; the mother’s
serum contains an IgG antibody of potential clinical significance and the father is, or
may be, heterozygous for the gene encoding the antigen of interest. Fetal genotyping
for RhD, C, c, E and Kell is available for suitable patients. A NZBS Transfusion Medicine
Specialist can be consulted on whether it is appropriate to refer samples for testing.
6.10 Intrauterine Transfusion (IUT)
Intrauterine red cell transfusion is used to correct fetal anaemia resulting from red cell
alloimmunisation (most commonly due to anti-D followed in order of importance by
anti-c and anti-K) and less commonly red cell aplasia due to fetal parvovirus infection.
Similarly, intrauterine platelet transfusions are used to correct fetal thrombocytopenia
due to platelet alloimmunisation. The goal of IUT is primarily to prevent (or treat) fetal
hydrops, ensuring the fetus reaches a viable gestational age and can be delivered.
Cellular components for IUT must be irradiated.
Consultation with a NZBS Transfusion Medicine Specialist is required to access these
components. The specification for the component will be agreed in advance with the
fetomaternal specialist and information on the actual composition of the component
provided at the time of delivery of the product.
The general specification for red cell and platelet components for use in IUT are as
follows:
Transfusion Medicine Handbook 3rd Edition
Page 123
Table 6.11: Component Requirements for IUT
Component
Requirements
Red cells1
■
Group O (low titre IgG anti-A, -B) or ABO identical with fetus (if
group known)
■
RhD negative (if HDFN due to anti-c or other antibodies, may be
RhD positive)
■
IAT crossmatch compatible with maternal plasma (antigen
negative for relevant antigen)
■
< 5 days old
■
CMV negative
■
Irradiated
■
Haematocrit 0.75 - 0.90
Platelets
■
Group O (low titre IgG anti-A, -B) or group specific/compatible with
maternal antibody
■
HPA-compatible with maternal antibody
■
Collected by apheresis where possible
■
CMV negative
■
Irradiated
■
Volume required (mL) = Desired plt increment x Feto-placental BV2
Plt count of concentrate
■
Concentrated to platelet count 2000 - 4000 x 109/L
■
Rate of transfusion 1 - 5 mL/min
1See Table 4.5: Red Cell Components Available from NZBS.
2Blood Volume (BV).
6.11 Transfusion of the Newborn
Red Cells
Normal blood volume at birth varies according to gestational age and timing of clamping
of the cord. For term infants the average blood volume is 80 mL/kg whilst in pre-term
infants it is higher at 106 mL/kg. The newborn bone marrow does not respond as
rapidly as an adult and any uncorrected blood loss can rapidly lead to anaemia.
A significant cause of anaemia in neonates is iatrogenic blood loss associated with
laboratory testing. Neonates are also at increased risk of infection and these factors
must influence transfusion medicine practice, especially in premature infants.
The volume of red cells to be transfused in newborns depends on the weight of the baby
and local policy. International guidelines recommend a volume for top-up transfusion
of 10 - 20 mL/kg (although some neonatal units use the range 8 - 12 mL/kg) at an
infusion rate of 5 mL/kg/hr. Frusemide given at a dose of 1 mg/kg halfway through
the transfusion may sometimes be required to minimise volume load.
In some centres it is practice to minimise the number of donors to which a newborn is
exposed. NZBS provides small volume ‘neonatal’ red cell units (each typically containing
70 mL) which are produced by separating an adult ‘plasma-reduced’ red cell donation
into a number of equal-sized aliquots. All (or some) of the individual aliquots from one
Page 124
Transfusion Medicine Handbook 3rd Edition
adult unit can be reserved for sequential transfusions to the same baby over the full
35-day shelf life of the red cells. Fresh frozen plasma and platelet concentrates are
also available in neonatal volumes.
Compatibility Testing
The normal rules for ABO and RhD compatibility apply. However, for infants up to the
age of 4 months, maternal blood may be used for pretransfusion testing provided the
ABO groups of mother and infant are compatible since any clinically significant red
cell antibody present in the infant’s circulation will have come from the mother. Blood
components selected for transfusion should be compatible with any ABO or other red
cell antibody present in the maternal or infant plasma. Repeated transfusions during
the infant’s first 4 months of life do not require further compatibility testing if there are
no atypical maternal red cell antibodies in the maternal or infant plasma. However,
if the antibody screen is positive, full pretransfusion testing and crossmatching will
be necessary.
Pretransfusion testing of infants 4 months or older should be performed on a sample
from the infant.
Exchange Transfusion
For severe cases of HDFN, neonatal exchange transfusion may be required.
Table 6.12: Red Cell Component Requirements for Neonatal Exchange Transfusion
Red Cell Requirements For Exchange Transfusion1
■
Group O or ABO compatible with maternal and infant plasma
■
RhD negative (or RhD identical with the infant)
■
Negative for red cell antigens against which maternal antibody is directed
■
IAT crossmatch compatible with maternal plasma
■
< 5 days old
■
CMV negative
■
Irradiated, especially if infant has previously received IUT
■
Haematocrit 0.5 - 0.60
■
Transfusion volume: 80 - 160 mL/kg for term infants and 100 - 200 mL/kg for
preterm infants, guided by clinical indication
1See Table 4.5: Red Cell Components Available from NZBS.
Special Requirements
Neonates less than 1500 g in weight or immunosuppressed for other reasons should
be given ‘CMV safer’ blood components as an added precaution. The use of either
prestorage leucodepletion or selection of CMV-antibody negative blood components
significantly reduces the risk of CMV transmission and CMV disease in susceptible
recipients. However, neither method alone or in combination can completely avoid
transmission from the occasional donor with CMV viraemia in the “window” period
prior to the development of antibodies following acute infection or when reactivation
of latent infection occurs.
Transfusion Medicine Handbook 3rd Edition
Page 125
In some situations irradiated blood components may be indicated for neonatal
transfusions. Irradiation removes the small risk of transfusion-associated graft-vs-host
disease (TA-GVHD) in at-risk patients by inactivating donor lymphocytes.
When there is clinical suspicion of a congenital cellular immunodeficiency state such as
severe combined immunodeficiency, DiGeorge syndrome, Wiskott-Aldrich syndrome,
ataxia telangiectasia or partial deletion of chromosome 22 (del 22q11.2), use of
irradiated components is mandatory. Features associated with immunodeficiency
states include cardiac defects, dysmorphic features, craniofacial abnormalities,
hypocalcaemia and lymphopenia.
Irradiated components are not indicated for ‘top-up’ transfusions of premature or term
infants unless there has been a previous IUT, in which case irradiated components
should be administered until 6 months after the 40 week gestation date.
Irradiated components may also be appropriate for extreme low body weight neonates
less than 28 weeks who are at increased risk of TA-GVHD due to physiological
immune incompetence. Evidence suggests that the risk is greatest in those < 900 g
in body weight.
As a result of gamma irradiation, red cells lose potassium during storage. While this
is not normally a problem with small volume neonatal top-up transfusions, for larger
volume and exchange transfusions irradiated red cells must be transfused within 24
hours of irradiation.
Use of Erythropoeitin
In some infants erythropoietin (EPO) may be used to reduce transfusion requirements.
Specialist advice on dose and frequency should be obtained.
Platelets
There is an increased risk of haemorrhage in preterm infants with moderate
thrombocytopenia < 50 x 109/L and in full-term infants with platelet counts less
than 20 - 30 x 109/L. The risk is increased if sepsis or coagulopathy are present.
The recommended volume for top-up transfusion is 10 mL/kg to a maximum of
one neonatal unit. A single neonatal platelet unit will normally provide an acceptable
platelet increment in children under 10 kg in body weight. At higher body weights, one
single neonatal platelet unit per 10 kg of body weight followed by a post-transfusion
platelet count and reassessment of the patient is recommended. If the plasma volume
of the platelet concentrate is excessive, the blood bank may be asked in advance
for advice regarding the need to remove plasma to a minimum volume of 10 - 15 mL
per platelet concentrate.
6.12 Neonatal Autoimmune Thrombocytopenia
Neonates with thrombocytopenia associated with maternal autoimmune disease (e.g.,
immune thrombocytopenia or systemic lupus erythematosis) generally have a benign
postnatal course without bleeding complications and one in which often a nadir is
reached by day 2 - 3, followed by a spontaneous rise in platelet count by the seventh
day. In a small number of cases treatment is warranted due to persistent severe
thrombocytopenia. Most neonates respond well to intravenous immunoglobulin in a
dose of 2 g/kg body weight. Platelet transfusions have no value in prophylaxis of this
condition but may be useful if there is bleeding.
Page 126
Transfusion Medicine Handbook 3rd Edition
6.13 Fetal and Neonatal Alloimmune Thrombocytopenia (FNAIT)
FNAIT is a rare but serious condition which arises from maternal alloimmunisation
to fetal platelet antigens of paternal origin and which affects approximately 1:1,100
pregnancies. FNAIT is the platelet equivalent of HDFN, where maternal IgG antibodies
cross the placenta and may destroy fetal platelets, leading to thrombocytopenia and
an increased risk of bleeding. The most serious consequence, estimated to occur
in 10 - 20% of untreated pregnancies and presenting at any time from 20 weeks
gestation until a few days after birth, is intracranial haemorrhage (ICH) which may be
fatal or lead to long-term neurological problems.
The most common alloantibody responsible for FNAIT in Caucasians is anti-HPA-1a
(80 - 90% of cases) followed by anti-HPA-5b (5 - 15%), with other HPA antibody
specificities are infrequently seen. In Maori and Asian mothers, consideration should
be given to other antibodies including those within the HPA-4 system.
There may or may not be a history of thrombocytopenia in a previous infant. Unlike
HDFN, FNAIT can occur during a first pregnancy and in fact almost half of the clinical y
evident cases are discovered in the first live-born infant.
Treatment
The management of FNAIT is overseen by a fetal medicine specialist along with input
from a specialist haematologist and/or NZBS Transfusion Medicine Specialist.
In the neonatal period the condition is self-limiting often resolving within two weeks after
birth although occasionally persisting for up to eight weeks. The treatment of choice
is platelet transfusion for which platelets lacking the specific HPA antigen should be
selected. In the absence of suitable donor platelets, the mother’s platelets may be
used. These must be washed to remove the plasma that contains the platelet antibody
and must be irradiated. Such a selected donation must follow the usual procedures
for collecting, testing, storing, handling and transfusing of the unit that apply to non-
selected blood donations.
For well neonates with suspected FNAIT, no evidence of haemorrhage, and where
the platelet count is < 30 x 109/L, prompt transfusion of platelets negative for both
HPA-1a and HPA-5b antigens is recommended. Following the results of serological
testing, directed donations negative for the implicated antigen are recommended.
Where there is evidence of ICH or other major haemorrhage, a higher platelet threshold
50 - 100 x 109/L should be used. The platelet count should be maintained > 50 x
109/L at least until the end of the first week of life. In view of the poor outcome of
FNAIT-associated ICH, if HPA antigen-negative or washed maternal platelets cannot be
provided, current BCSH guidelines recommend the transfusion of randomly selected
platelets (irrespective of their HPA status). Intravenous immunoglobulin (IVIg) is an
alternative therapy, although there is often a 24 to 48 hour delay in response, and may
be used in combination with platelet transfusion. Treatment of the neonate with high
dose intravenous immunoglobulin (2 g/kg body weight) is effective in about 65% of
cases and can reduce the period of dependence on compatible platelets.
For women with a history of FNAIT, management of the fetus during a subsequent
pregnancy includes assessment of ICH risk (including paternal HPA zygosity +/- fetal
HPA genotyping), antenatal administration of IVIg 1 - 2 g/kg/week from as early as
12 weeks, and/or corticosteroids. Despite immune modulating therapy, there remains
Transfusion Medicine Handbook 3rd Edition
Page 127
an increased risk of fetal ICH. For cases considered to be at high risk of ICH (such
as a previous child with FNAIT-associated ICH), prophylactic intrauterine fetal platelet
transfusions may be included in the management.
6.14 Individuals Refusing Blood Transfusion
Some patients may refuse to receive transfusion for personal, religious or cultural
reasons. For example, Jehovah’s Witnesses may hold beliefs that preclude transfusion
of blood components or fractionated products. These wishes and beliefs must be
respected and patients treated in a non-emotional and logical way.
Some patients will refuse all blood transfusions while others will accept fractionated
products. Others will accept autologous blood collection and transfusion, cell salvage
and auto-transfusion of blood collected during surgery. Some patients do not wish
their relatives or peers to know that they have been transfused.
When a patient indicates that they do not wish to receive a transfusion there should be
close and confidential discussion with the patient as to their exact requirements and
what they will and will not accept. This information should be recorded in their notes.
It is advisable that the matter is discussed with the clinical leader of the unit concerned.
Where there are areas of difficulty or dissension, the Chief Medical Officer (CMO) of
the hospital should be contacted for further advice and guidance as to management
of the patient. The CMO will have access to the DHB solicitor and appropriate patient
advocate groups. Additional consultation may be necessary with anaesthetists and
surgeons to ensure the best possible care is given to the patient when blood transfusion
cannot be provided despite clear clinical indication.
For children, the same procedure should be followed with careful consultation of the
parents and noting any comments or opinions from the child. For babies and infants,
the clinical leader should be closely involved in the decision-making. In cases where
the parents, whanau or guardians refuse consent for transfusion deemed necessary to
save life, prevent permanent injury or prolonged and avoidable pain and suffering, the
consultant responsible needs to consult with the CMO to discuss appropriate further
action. Such action may involve meeting with a solicitor to discuss the need for legal
action. If court action is deemed necessary, an
inter partes hearing may be appropriate.
It is mandatory that a very careful procedure be followed, with full documentation and
consultation with the relevant people, prior to any transfusion.
Page 128
Transfusion Medicine Handbook 3rd Edition
7
ADVERSE EFFECTS OF TRANSFUSION
7.1 Overview
Transfusion, like other treatments, can both benefit and harm the patient. Good clinical
practice depends on understanding both the benefits and risks that transfusion may
carry. Also, it is essential to consider the potential benefits and risks of not using a
blood component or fractionated product, or of using an alternative.
Blood transfusion has become safer as infectious hazards have been identified and
donor selection procedures, viral screening tests and platelet pre-release bacterial
culture systems have been introduced. There has also been continuous improvement
in manufacturing processes for fractionated products.
Although much effort is placed into ensuring the safety of transfusion, preventable
clerical error and inappropriate transfusion still account for a significant proportion of
reported transfusion-related adverse events.
Data on the occurrence of adverse reactions and other adverse events associated
with the transfusion process are collected through NZBS Haemovigilance.
7.2
Reporting Adverse Reactions and Events
Serious or life-threatening acute reactions are rare but new or unexpected symptoms
that appear while the patient is being transfused must not be overlooked, as they may
be early warning signs of a serious reaction.
Severe reactions are most likely to occur within 15 minutes of starting a transfusion.
It is important to monitor the patient closely during this initial period and thereafter at
regular intervals according to local hospital policy. Serious events should be discussed
with a NZBS Transfusion Medicine Specialist/Medical Officer or specialist haematologist
for advice on further management of the patient, laboratory investigations and future
transfusion requirements.
Adverse reactions to blood components
If the patient experiences an adverse reaction during or following transfusion of a blood
component, clinical staff must report this to the blood bank as soon as possible. NZBS
supplies a
Transfusion-related Adverse Reaction Notification form that has guidelines
for the management of adverse transfusion reactions to assist clinical staff in the
immediate care of the patient.
The completed form should be sent to the blood bank along with remnants of the
transfused components and laboratory samples required for full investigation of the
reaction. Refer to Table 10.1:
Samples Required for Pretransfusion Testing for further
information.
Adverse reactions to fractionated products
If the patient experiences an adverse reaction to a fractionated plasma product this
must be reported to the blood bank as soon as possible, for example by sending a
NZBS
Transfusion-related Adverse Reaction Notification form. The blood bank will
then initiate completion of a NZBS
Notification of Adverse Reaction to a Fractionated
Product form together with information obtained from the patient’s clinician.
Transfusion Medicine Handbook 3rd Edition
Page 129
In accordance with pharmacovigilance requirements, NZBS reports the occurrence of
adverse reactions to fractionated products to the relevant manufacturer, for example
CSL Behring. Where clusters of similar adverse reactions occur, these are reported
to the New Zealand regulator, Medsafe. Both Medsafe and the Centre for Adverse
Reactions Monitoring (CARM) are provided with an annual NZBS Haemovigilance
Report.
Adverse events associated with transfusion
The blood bank should be notified as soon as possible if it is believed that the patient
has received a wrong blood component or fractionated product, received one
intended for another patient, that the transfusion did not meet requirements or that
the transfusion was inappropriate. It should be noted that the patient may not always
experience or show a ‘reaction’ in these situations.
Page 130
Transfusion Medicine Handbook 3rd Edition
eatment for
eaction is mild and
opriate tr
ecipient identity
e not appr
etic Paracetamol 1g po and monitor
oids ar
Check label and r
Slow the transfusion if r
MO elects to continue transfusion
Send Haemovigilance notification to Blood
Bank
Antipyr
closely
Ster
FNHTR
Management
■
■
■
■
■
etic
e
ent
ecurr
e
oduction and
epeated minor
e r
ed
ed
evious transfusion
etransfusion antipyr
evention
Check for history of
pr reactions. Consider pr Paracetamol 1g po wher reactions occur and further transfusions ar requir
Consult TMS if r
reactions occur
Note: All blood
components ar depleted of leucocytes during pr further leucodepletion at the bedside is not requir
Pr
■
■
■
ransfusion Reactions
ransfusion Reactions
,
e rise
e
e rise
esent:
<1.5°C
espiratory
etransfusion
unexplained
e rise of at least
om pr
dia, headache,
etransfusion baseline,
unexplained fever
unexplained fever
e:
ess and haemodynamic
≥38°C and a
ehension, loin pain,
Mild: ≥38°C and a temperatur of at least 1°C but from pr occurring in the absence of chills, rigors, r distr instability
Moderate: fever temperatur 1°C but not meeting criteria for either mild or sever FNHTR
Sever >39°C and a temperatur ≥2.0°C fr baseline and chills/rigors Associated or secondary symptoms may be pr tachycar nausea, flushing, anxiety hypertension or occasionally hypotension In some cases: marked appr and/or angina
Signs & Symptoms
■
■
■
■
■
reaction
1-3:100
Guidelines for the Management of Adverse T
Reaction induced by cytokines
Alloimmune to leucocyte antigens
Other causes may exist
Common
Onset during or within
4 hours following transfusion
equency:
7.3
Table 7.1: Guidelines for the Management of Adverse T
Reaction/Cause
Febrile Non-Haemolytic Transfusion Reaction (FNHTR)
Fr
(higher in multi-transfused
recipients)
■
■
Transfusion Medicine Handbook 3rd Edition
Page 131
e
eat as a moderate or
ecipient identity
ease tr
eaction
esume
e r
eased monitoring, e.g., BP/TPR every 15-
oublesome
Slow transfusion
Check label and r
Antihistamine, e.g., Loratadine 10mg
or Cetirizine 10mg po if symptoms ar
tr
If symptoms mild and transient, transfusion
may r
Continue transfusion at a slower rate with
incr
30 min
Send Haemovigilance notification to Blood
Bank
If symptoms incr
sever
Management
■
■
■
■
■
■
■
e
ophylaxis
ent mild
ophylaxis
ecipients befor
ecurr
evention
For r
reactions pr
with antihistamine to
alleviate symptoms, e.g.,
Loratadine 10mg or
Cetirizine 10mg po
Routine pr
for all r
transfusion is not
indicated
Pr
■
■
ransfusion Reactions continued
eaction:
Flushed skin
Morbilliform rash with itching
Urticaria (hives)
Localised angioedema
Periorbital itch, erythema
and oedema
Conjunctival oedema
Minor oedema of lips,
tongue and uvula
Signs & Symptoms
Minor or localised r
■
■
■
■
■
■
■
otein
oduct
eacting
1:100 – 1:500
e common with
Recipient may have
an antibody r
with plasma pr
or leucocyte antigen
(HLA or other) in
the transfused
component or
fractionated pr
Mor
plasma and platelet
components
Onset during or within
4 hours following
transfusion
equency:
Reaction/Cause
Allergic Reaction (minor)
Fr
■
■
Table 7.1: Guidelines for the Management of Adverse T
Page 132
Transfusion Medicine Handbook 3rd Edition
ocortisone may be
eat symptomatically
omethazine 25-50mg IV
eactions with TMS
ecipient identity
ed with IV fluids, oxygen and
ed
equir
Stop transfusion
Check label and r
Replace IV set and give saline to keep vein
open and/or maintain BP
Monitor closely and tr
as r
antihistamine, e.g., Pr
(max rate 25mg/min) or Loratadine 10mg or
Cetirizine 10mg po. Hydr
consider
Send Haemovigilance notification to Blood
Bank
Discuss moderate r
Management
■
■
■
■
■
■
e
event
ophylactic
eactions but
esent
ent
ocortisone will
eliably pr
eatment with an
evention
Discuss with TMS if
recurr
Note: Pr
tr
antihistamine or
hydr
not r
moderate and sever
allergic r
may alleviate symptoms
when pr
Pr
■
■
ransfusion Reactions continued
ead
e common
dia
e anxiety
Symptoms as for minor
reactions, plus:
Cough
Mild hypotension or
tachycar
Dyspnoea and oxygen
desaturation ar
Chills and rigors
Loin pain and angina
Sever
Signs & Symptoms
Moderate or widespr
reaction:
■
■
■
■
■
■
■
oduct
may
otein or
1:500 –
1:5,000
Recipient
have an antibody
reacting with a
plasma pr
leucocyte antigen
(HLA or other) in
the transfused
component or
fractionated pr
Onset usually within
first 50-100mL infused
and within 4 hours of
transfusion
equency:
Reaction/Cause
Allergic Reaction
(moderate)
Fr
■
Table 7.1: Guidelines for the Management of Adverse T
Transfusion Medicine Handbook 3rd Edition
Page 133
esent and eatment
ns to normal
en 20mL/kg, until
etur
epeat at 5-10 min
esent (r
eactions
e r
e contraindicated
ecipient identity
ed:
ocortisone 4mg/kg (200-400mg
eactions with TMS
equir
e r
0.5mg/0.5mL
en 0.01mg/kg IM; min dose 0.1mL,
hours)
>90mmHg, then titrate
enalin 1:1000 IM and r
Adult:
Childr max dose 0.5mL
omethazine ar
Adr intervals if r
saline, e.g., adults 2L, childr SBP
IV)
Cetirizine 10mg po for itch or angioedema
added for sever
Pr
AP ventilation, chest x-ray
Stop transfusion
Check label and r
Follow Anaphylaxis Guidelines:
•
• Replace IV set and give rapid IV colloid or
• Consider Hydr
• Consider H1-antihistamine, e.g., Loratadine or
• H2-antihistamine, e.g., Ranitidine may be
• Note: Sedating antihistamines, e.g.,
CP
ICU liaison
Follow NZRC Guidelines if no pulse pr
for symptoms that persist after initial tr
Collect serum tryptase sample within 1-2 hours
if anaphylaxis may be pr within 6-8
Send Haemovigilance notification to Blood Bank
Discuss sever
Management
■
■
■
■
■
■
■
■
■
e
ecipient is
e the
eaction is
oducts may be
opriate if r
ashed cellular
evention
IgA deficient blood/
blood pr appr known to have absolute IgA deficiency and to have anti-IgA
W
components may be indicated wher cause of the r not identified
Pr
Discuss with TMS befor requesting:
■
■
ransfusion Reactions continued
, change in
eaction:
ecipients
dia
ead urticaria with
e hypotension, shock
e anxiety
eatening r
idespr
Symptoms as for moderate
reactions, plus:
Sever
and tachycar
W
skin flushing and itching
Wheezing, stridor
voice
Sever
Note: Respiratory
symptoms may dominate in anaesthetised r
Signs & Symptoms
Life-thr
■
■
■
■
■
■
e)
may
otein
otein
1:20,000 – 1:50,000
oduct, e.g., IgA,
Recipient have an antibody reacting with a plasma pr in the transfused component or fractionated pr haptoglobin or other plasma pr
Rapid onset
equency:
Reaction/Cause
Anaphylactic / Anaphylactoid Allergic Reaction (sever
Fr
■
Table 7.1: Guidelines for the Management of Adverse T
Page 134
Transfusion Medicine Handbook 3rd Edition
esolved
Stop transfusion
Replace the IV infusion set and infuse saline
to manage BP
Symptomatic management until r
Send Haemovigilance notification to Blood
Bank
Management
■
■
■
■
evention
Pr
ransfusion Reactions continued
systolic BP
and
hour of completing
>30mmHg during or
Hypotension – fall in systolic
BP
within 1
transfusion
<80mmHg
Signs & Symptoms
■
eased
incr
1-2:1,000
idiosyncratic
Often
reaction
Possible
risk with ACE-
inhibitor therapy
Onset during or within 1
hour of transfusion
equency:
Reaction/Cause
Hypotensive Reaction
Fr
■
Table 7.1: Guidelines for the Management of Adverse T
Transfusion Medicine Handbook 3rd Edition
Page 135
e with
essur
ed
eat if clinically
enal and liver function,
ecipient identify
etic, e.g., Frusemide 1-2mg/kg IV and/or
ocortisone may be consider
Stop transfusion
Check label and r
Replace IV set and start normal saline
Treat shock and maintain blood pr
IV saline infusion
Investigate possible DIC and tr
significant bleeding
Diur
Mannitol, may help maintain urine flow
Hydr
Samples to assess r
DIC and haemolysis, e.g., full blood count,
unconjugated bilirubin, LDH and haptoglobin
Send Haemovigilance notification to Blood
Bank
Management
■
■
■
■
■
■
■
■
■
s
s ID
ecipient’
ecipient of
ecipient’
eful monitoring of
e and handle blood
etransfusion blood
oper administration of
evention
Meticulous checking
of r
and labelling of
pr
sample at r
side
Meticulous two-person
checking of ID of
intended r
blood component and
component label
Car
recipient for first 15 min
of each unit transfused
Stor
components within
specifications
Pr
blood components
Pr
■
■
■
■
■
ransfusion Reactions continued
Co
>1
dia
, jaundice
haemoglobinuria
Unexplained fever
Chills, rigors
Pain up arm
Chest, abdominal or low
back pain
Dyspnoea
Tachycar
Hypotension, shock
Haemoglobinaemia
and
Oliguria with dark urine or
anuria
Nausea, vomiting
Diarrhoea
Pallor
Bleeding (due to DIC)
Signs & Symptoms
Some or all of –
■
■
■
■
■
■
■
■
■
■
■
■
■
ed
ed
handling,
1:12,000 –
1:100,000
, ABO
ely
oper
ABO or other
incompatible r
cell transfusion
reaction caused by
complement-fixing
antibodies
Rar
antibodies in a
platelet or plasma
component
Impr
storage or
administration of r
cell component
Onset within 24 hours,
usually immediate
equency:
Reaction/Cause
Acute Haemolytic
Transfusion Reaction
Fr
■
Table 7.1: Guidelines for the Management of Adverse T
Page 136
Transfusion Medicine Handbook 3rd Edition
ed)
een (may be
ed)
ed cells clear
oup antibody scr
eaction is suspected
ed cells clear
ect antiglobulin test (may be negative when
Investigate haemolysis:
Full blood count with film comment
Dir
most r
Blood gr
negative until r
Liver function tests
Haptoglobin concentration falls while
haemolysis is occurring
LDH
Send Haemovigilance notification to Blood
Bank if r
Management
■
■
■
■
■
■
■
■
e
ed
oup antibodies
ded on the
ecor
ovided for futur
oviding compatible
e r
evention
Blood gr
ar
NZ Blood Service
national database so
that compatible r
cell components can
be pr
transfusions
Note: Delay may occur
in pr
red cells for transfusion
Pr
■
■
,
ransfusion Reactions continued
esent
om destruction of
een shows
ely splenomegaly
e cases
ocytes may be pr
orsening anaemia and W jaundice fr red cells Often asymptomatic but rar haemoglobinaemia and haemoglobinuria Renal impairment may occur in sever Blood scr unexpected anaemia and spher on film
Signs & Symptoms
■
■
■
■
esults
days
.
essing
eported
has
oup
oduces a
ed cells
egnancy
elevant
eviously been
Recipient
pr
sensitised to
a blood gr
antigen, usually
by transfusion
or pr
Transfusion with
red cells expr
the r
antigen pr
secondary immune
response and r
in haemolysis of
transfused antigen-
positive r
Onset usually 1-7 days
post-transfusion but
may be up to 28
equency:
Reaction/Cause
Delayed Haemolytic
Transfusion Reaction
Fr
Estimated 1:5,000 but
recognized and r
events 1:35,000
■
Table 7.1: Guidelines for the Management of Adverse T
Transfusion Medicine Handbook 3rd Edition
Page 137
or
penicillin
ecipient if sepsis
om r
es fr
oad-spectrum
br
a
es on blood component and
antibiotics:
Stop transfusion
Replace IV set; give saline to maintain BP
and/or keep vein open
Send Haemovigilance notification to Blood
Bank
Notify Blood Bank by phone and contact TMS
urgently
Obtain blood cultur
suspected
Give
cephalosporin and gentamicin 5mg/kg
Note: Blood Bank will arrange urgent Gram
stain and cultur
report any positive findings
Management
■
■
■
■
■
■
■
ed
e and handle
oducts for any
ed or leaking
e transfusing:
a visibly clumped
platelet component
an unusually dark r
cell component
punctur
bag
evention
Collect, stor
blood components
within specifications
Inspect pr
visual abnormality or
defect in unit container
befor
•
•
•
Pr
■
■
ransfusion Reactions continued
ed cell
ess,
Yersinia
sepsis
, chills, fever
Rigor
Shock, usually within
minutes of starting
transfusion
Respiratory distr
wheezing and oxygen
desaturation
Pain up arm
Chest and back / loin pain
Nausea, vomiting
Explosive diarrhoea
may occur with
enterocolitica
Most common infecting
agents: staphylococcal
species (platelet
components), gram
negative species (r
components)
Signs & Symptoms
■
■
■
■
■
■
■
■
ely
esent,
own to a
e pr
ed cells
component
commonly
fects platelet
fects r
Blood
contains bacteria
that have gr
high concentration
Most
af
components; rar
af
If gram negative
bacteria ar
endotoxin levels may
be very high
Rapid onset
equency:
Reaction/Cause
Bacterial Sepsis
Fr
Platelet components:
<1:10,000
Red cell components:
<1:250,000
■
Table 7.1: Guidelines for the Management of Adverse T
Page 138
Transfusion Medicine Handbook 3rd Edition
etic (Frusemide
AP ventilation
ecipient upright with legs over side of
Stop transfusion
Seek urgent medical assessment
Sit r
bed, administer oxygen, diur
1-2mg/kg IV), CP
Phlebotomy may be necessary
Send Haemovigilance notification to Blood
Bank
Management
■
■
■
■
■
ed
ecipient
eserve
in Units
ecipients
equir
not
etic
escribe
diovascular or
ed or a large
en, and r
opriate for r
diovascular r
void elective
evention
Restrictive transfusion
practice
Monitor fluid balance
esp. in elderly and
childr
with car
renal disease
Transfuse at a rate
appr
Give a diur
immediately prior
to a transfusion if
car
is impair
transfusion is r
A
transfusions at night
Always pr
paediatric transfusion
dose in mL,
Pr
■
■
■
■
■
■
e
ransfusion Reactions continued
ess
essur
diac silhouette
dia
espiratory distr
eased blood pr
oductive cough with pink
diac collapse
Acute r
along with some or all of
the following: dyspnoea,
oxygen desaturation,
pr
frothy sputum, nausea,
restlessness and anxiety
Tachycar
Incr
Acute or worsening
pulmonary oedema on chest
xray
Enlarged car
on chest xray
Evidence of fluid overload
such as positive fluid
balance, raised JVP and/or
CVP
Note: Hypotension may
occur in cases of acute
car
Signs & Symptoms
■
■
■
■
■
■
■
ed
culatory
ed cell
recipient
transfused
:
eat for
ecipient
ransfusion-
diovascular or
Elderly
with impair
car
renal function
Transfusion too rapid
for r
Volume
too gr
recipient, especially
if normovolaemic
Onset during or within
6 hours following
transfusion
Main risk factors:
•
•
•
equency
Reaction/Cause
TACO: T
Associated Cir
Overload
Fr
1:100 – 1:1,000 r
transfusion episodes
■
Table 7.1: Guidelines for the Management of Adverse T
Transfusion Medicine Handbook 3rd Edition
Page 139
e
ecipient
A antigen
elevant HP
ombocytopenia in the
eatments
ecommended
e thr
A antibodies
ombocytopenia or clinically
oids and plasma exchange ar
e thr
eatening bleeding – platelet
e desirable
void random-donor platelet transfusion
Consult TMS or haematologist if a r
of cellular blood components develops an
unexpected sever
following 1-2 weeks
Test for HP
If not bleeding – monitor
If sever
significant bleeding – intravenous
immunoglobulin is r
Corticoster
recognised additional tr
If life-thr
components lacking the r
ar
A
Management
■
■
■
■
■
■
■
e selected
oviding
omptly so that
equir
evention
Restrictive transfusion
practice
Notify Blood Bank and
TMS pr
relevant investigations
can be initiated
Further transfusions
may r
components
Note: Delay may
occur in pr
further cellular blood
components
Pr
■
■
■
■
ransfusion Reactions continued
ombocytopenia
e thr
ombocytopenia will
Sever
often with purpura and
possibly other bleeding
Thr
persist for 1-2 weeks
Signs & Symptoms
■
■
A). The
has
culating
oduced an
Recipient
pr
antibody to a human
platelet-specific
antigen (HP
antibody forms
immune complexes
with transfused
platelet antigens
resulting in clearance
of most cir
platelets
Onset about 5-12
days after transfusion
of cellular blood
components
equency:
egnant)
Reaction/Cause
Post-transfusion Purpura
Fr
<1:100,000 (mostly occurs in women who have been pr
■
Table 7.1: Guidelines for the Management of Adverse T
Page 140
Transfusion Medicine Handbook 3rd Edition
e
ecipient
A antigen
ed
espiratory
equir
elevant HP
ombocytopenia in the
eatments
ecommended
e thr
e management for r
A antibodies
ombocytopenia or clinically
oids and plasma exchange ar
e not usually helpful
e thr
eatening bleeding – platelet
e
etics ar
e desirable
void random-donor platelet transfusion
Consult TMS or haematologist if a r
of cellular blood components develops an
unexpected sever
following 1-2 weeks
Test for HP
If not bleeding – monitor
If sever
significant bleeding – intravenous
immunoglobulin is r
Corticoster
recognised additional tr
If life-thr
components lacking the r
ar
A
Intensive car
failur
Diur
Send Haemovigilance notification to Blood
Bank
Notify Blood Bank by phone and contact TMS
urgently
Tissue typing samples will be r
Management
■
■
■
■
■
■
■
Management
■
■
■
■
■
e
AS)
testing
om FFP
om the
e selected
oviding
educed by
esis platelet
omptly so that
equir
Male-only FFP
HLA-antibody
of apher
donors
Pooled platelets ar
suspended in platelet
additive solution (P
and have minimal
residual plasma
evention
Restrictive transfusion
practice
Notify Blood Bank and
TMS pr
relevant investigations
can be initiated
Further transfusions
may r
components
Note: Delay may
occur in pr
further cellular blood
components
evention
Restrictive transfusion
practice
NZ case-rate fr
and platelet components
has been r
supply of:
•
•
•
Notify Blood Bank
so that donor(s) can
be assessed for
relevant antibodies and
implicated donor(s)
withdrawn fr
active donor panel
Pr
■
■
■
■
Pr
■
■
■
ransfusion Reactions continued
ess
f
oductive
e) along
om:
ombocytopenia
espiratory distr
oceeding to
eaction occurs
e thr
ombocytopenia will
culatory overload)
TACO
other causes of acute
lung injury (ALI)
Sever
often with purpura and
possibly other bleeding
Thr
persist for 1-2 weeks
Acute r
(often pr
respiratory failur
with some or all of the
following: dyspnoea, oxygen
desaturation, non-pr
cough, chills, fever and
hypo- or hypertension.
Bilateral infiltrates on chest
xray
Absence of left atrial
hypertension (as in
cir
If the r
during anaesthesia the
lungs become very stif
from rapidly developing
pulmonary exudate
Distinguish fr
•
•
Signs & Symptoms
■
■
Signs & Symptoms
■
■
■
■
■
esults
oup
ecipient
e
A). The
has
culating
om ARDS
ders
<1:5,000
contributing
recognised
ophil (HNA)
ovascular lung
oduced an
ransfusion-
Recipient
pr
antibody to a human
platelet-specific
antigen (HP
antibody forms
immune complexes
with transfused
platelet antigens
resulting in clearance
of most cir
platelets
A complex gr
of disor
indistinguishable
clinically fr
One
mechanism involves
a donor antibody
reacting with r
leucocyte (HLA) or
neutr
antigens causing cell
activation that r
in acute sever
micr
injury
Other
factors likely exist
Onset about 5-12
days after transfusion
of cellular blood
components
Onset within 6 hours
following transfusion
of plasma or plasma-
containing cellular
components
equency:
egnant)
equency:
Reaction/Cause
Post-transfusion Purpura
Fr
<1:100,000 (mostly occurs in women who have been pr
■
Reaction/Cause
TRALI: T
Related Acute Lung
Injury
Fr
■
Table 7.1: Guidelines for the Management of Adverse T
Transfusion Medicine Handbook 3rd Edition
Page 141
Consult TMS or haematologist to investigate
and establish diagnosis
Send Haemovigilance notification to Blood
Bank
Management
■
■
esidual
otocol to
equiring
s transfusion
d
evention
Irradiate cellular
blood components
to inactivate r
lymphocytes
When notified of
a patient r
irradiation of cellular
components, NZBS
attaches a pr
the patient’
recor
Pr
■
■
ransfusion Reactions continued
ome with
, rash, liver dysfunction,
Clinical syndr
fever
diarrhoea and pancytopenia
Signs & Symptoms
■
cellular
and
elative
lymphoma
donation
nucleoside
A-GVHD)
esis platelets
ected
ersus-Host
ell-defined risk factors
Congenital immune deficiency
Intrauterine transfusion and neonatal exchange transfusion
Hodgkin
Autologous allogeneic HSCT
Dir from blood r
HLA-matched apher
Granulocyte transfusion
Purine analogue therapy
Alemtuzumab therapy
Onset 1-6 weeks
following transfusion
W include:
•
•
•
•
•
•
•
•
•
equency: e but fatal
Reaction/Cause
Transfusion-Associated Graft-V Disease (T
Fr Rar
■
Table 7.1: Guidelines for the Management of Adverse T
Page 142
Transfusion Medicine Handbook 3rd Edition
e
ed, infuse
ement
equir
ecipient and monitor
e measur
om the r
e temperatur
es cor
ough a warmer
Limit heat loss fr
BP/TPR
If further blood components r
thr
Note: Reliable determination of temperatur
requir
Management
■
■
■
s
e the
e is
oduce
er’
ough a
e will pr
ect temperatur
operly maintained and
evention
Give large fluid
infusions thr
warmer designed for
rapid infusion of blood
components and follow
the manufactur
instructions
Equipment must be
pr
validated to ensur
corr
achieved as excessive
temperatur
haemolysis
Pr
■
■
ransfusion Reactions continued
e
opic
egularity
ed platelet function
diac rhythm irr
fect
Reduced temperatur
May be associated with
car
and a negative inotr
ef
Impair
and coagulation
Signs & Symptoms
■
■
■
en)
e than
essive onset
ogr
oducts (mor
Pr
during rapid infusion of
large volumes of cold
fluids, including blood
pr
50mL/kg/h in adults or
15mL/kg/h in childr
equency: no data
Reaction/Cause
Cooling
Fr
■
Table 7.1: Guidelines for the Management of Adverse T
Transfusion Medicine Handbook 3rd Edition
Page 143
7.4
Febrile Non-Haemolytic Transfusion Reaction
Fever or rigors during red cell transfusion affect 1 - 3% of recipients and in the past
were usually attributed to the transfusion of white cells present in blood components.
Febrile non-haemolytic transfusion reactions (FNHTR) generally occur more frequently in
patients who have been alloimmunised to leucocyte antigens as a result of pregnancy
or recurrent transfusion. The use of leucocyte-depleted blood components has
undoubtedly reduced the occurrence of FNHTR, however the relatively large number
of reactions still seen suggests the involvement of other mechanisms and risk factors.
Febrile reactions during platelet transfusion have been attributed to leucocyte- and
platelet-derived cytokines that accumulate in the product during storage.
Classical symptoms of FNHTR are shivering, usually 30 - 60 minutes after the start
of the transfusion, followed by fever. Most reactions can be managed by slowing
or stopping the transfusion and giving an antipyretic such as paracetamol. FNHTR,
although unpleasant, are not life-threatening, however it is important to remember fever
or rigors can also be the early symptoms of a severe acute haemolytic transfusion
reaction or transfusion of bacterially contaminated blood.
Recurrent severe FNHTR in patients who require repeated transfusion of red cells or
platelets may be prevented by the use of washed cellular components.
Premedication
While treatment of FNHTR with antipyretics such as paracetamol for a symptomatic
rise in temperature may be justified, routine premedication with antipyretics and/or
antihistamines prior to transfusion is not advised as it is both unnecessary and may
modify important signs of a transfusion reaction. Steroids are not appropriate for the
treatment or prevention of FNHTR.
7.5
Allergic & Anaphylactic Reaction
Allergic reactions represent a spectrum of severity from mild, where the patient simply
experiences isolated urticaria or a rash, though to fatal anaphylactic shock.
Allergic reaction
These are typified by one or more of the following: urticaria, rash, allergic dyspnoea
(stridor, cyanosis, wheezing), localised angioedema or generalised pruritis without
hypotension during or within 4 hours of transfusion. These reactions are commonly
associated with transfusion of components with large volumes of plasma, for example
platelets and FFP. Since the introduction of platelets suspended in platelet additive
solution (PAS), the frequency of allergic reactions has reduced.
Symptoms usually subside if the transfusion is slowed and parenteral antihistamine
is given. The transfusion may be continued if there is no progression of symptoms
after 30 minutes.
A rise of mast cell tryptase can support the diagnosis of an allergic reaction.
Anaphylactic reaction
These are rare but life-threatening complications usually occurring during or very
shortly after transfusion and are differentiated from mild/moderate allergic reactions
by severity where, in addition to mucocutaneous features, there is airway compromise
Page 144
Transfusion Medicine Handbook 3rd Edition
or severe hypotension requiring vasopressor treatment (or associated symptoms like
hypotonia or syncope).
Anaphylaxis may occasionally be associated with antibodies against IgA in patients
who have extremely low levels of IgA in their plasma or other genetic variants of
plasma proteins. If this is the suspected cause the patient should, if possible, not
be transfused. Special components will be needed in consultation with an NZBS
Transfusion Medicine Specialist.
Premedication
Treatment with an antihistamine or hydrocortisone for generalised allergic reactions
is justified. Premedication may be appropriate before transfusing a patient who has
previously experienced repeated allergic reactions. Routine premedication with
antihistamines prior to transfusion is however not advised, as it is both unnecessary
and may modify important signs of a transfusion reaction.
7.6
Hypotensive Transfusion Reaction
Hypotensive transfusion reactions are defined as a drop in systolic blood pressure
≥ 30 mmHg during or within one hour of transfusion together with a systolic blood
pressure ≤ 80 mm Hg. Most reactions occur very rapidly within minutes of starting
the transfusion and respond to ceasing the transfusion together with supportive care.
The underlying condition of the patient must have been excluded as an explanation
for the reaction. These may occur more frequently in patients receiving angiotensin-
converting enzyme (ACE) inhibitor therapy.
7.7
Acute Haemolytic Transfusion Reaction
Incompatible transfused red cells react with the patient’s anti-A or anti-B antibodies and
cause an acute haemolytic transfusion reaction (AHTR). Such a reaction can activate
complement and cause disseminated intravascular coagulation (DIC) and acute renal
failure. The reaction is usually most severe if group A red cells are transfused to a
group O patient. Transfusion of ABO-incompatible blood almost always arises from
pretransfusion sample labelling errors or from failure to perform required checks prior
to giving the transfusion. If red cells are administered to the wrong patient (i.e., any
patient other than the one for whom the red cells were supplied) the chances of ABO-
incompatibility are about 1 in 3. Rarely, AHTR is due to a non-A, non-B, complement-
fixing antibody. Such reactions reported most commonly involve the Kell, Duffy and
Kidd antigen group systems.
Acute haemolysis may also occur following transfusion of plasma-rich blood
components such as platelets or FFP from donors with high titres of anti-A or anti-B
that react with patient red cells.
In a conscious patient even a few mil ilitres of incompatible blood may cause symptoms
within a few minutes of starting the transfusion. The patient may become restless or
distressed and experience pain at the infusion site, fever, flushing, breathlessness, or
abdominal, flank or substernal chest pain. The severity varies widely as it is dependent
on the titre of blood group antibody in the recipient, the quantity of blood transfused
and other factors such as age. In an unconscious or anaesthetised patient, hypotension
and uncontrollable bleeding due to DIC may be the only signs of an incompatible
transfusion. Oliguria is common and is often followed by acute renal failure.
Transfusion Medicine Handbook 3rd Edition
Page 145
If AHTR is suspected, the transfusion must be stopped, the line maintained with
intravenous saline and urgent steps taken to confirm or exclude this possibility.
Signs and symptoms of AHTR may be similar to a severe allergic reaction or bacterial
contamination. In addition, autoimmune haemolytic anaemia due to erythrocyte
autoantibodies in the recipient and non-immune (mechanical) causes must be part
of the differential diagnosis.
7.8
Delayed Haemolytic Transfusion Reaction
A delayed haemolytic transfusion reaction (DHTR) is one in which evidence of increased
red cell destruction develops, usually between 24 hours and 28 days, following a
transfusion. The symptoms and clinical or laboratory signs are similar to AHTR but are
usually less severe, often manifesting as an inadequate rise or unexplained fall in the
post-transfusion haemoglobin. Whilst clinically significant DHTR are rare and seldom
fatal, they can cause additional problems for a patient who is already seriously ill.
DHTR occur in a patient immunised to a red cell antigen by an earlier transfusion
or pregnancy. The level of antibody may be so low that it cannot be detected in the
pretransfusion sample. After transfusion of red cells bearing the target antigen, a
rapid secondary immune response boosts the antibody level so that, after a few days,
transfused red cel s bearing the relevant antigen may be rapidly destroyed. Antibodies
of the Kidd and Rh systems are the most frequent cause of DHTR.
7.9
Bacterial Sepsis
Bacterial sepsis, whilst rare, is the leading microbial cause of transfusion mortality.
Sources of bacteria in blood components may include contamination from skin
organisms at the phlebotomy site due to ineffective skin disinfection, skin plugs
introduced to units during phlebotomy, transient bacteremia in donors and, rarely,
contamination during handling and processing of components.
Bacterial contamination is more likely in components stored at room temperature (20
- 24°C) such as platelets, than with red cells (stored at 2 - 6°C). Common organisms
associated with contamination include
Staphylococcus epidermidis, Staphylococcus
aureus, Bacillus cereus, Group B streptococci,
Escherichia coli, Pseudomonas species
and other gram-negative organisms. Sepsis due to contaminated platelets is thought
to be both under recognised and under reported. Platelet-associated sepsis is not
normally catastrophic and can occur several hours or longer post-transfusion making
it difficult to associate with transfusion and thereby diagnose. This is in contrast to the
sepsis and toxaemia from bacterially contaminated red cells which is often rapid and
catastrophic, with reported mortality rates of up to 60%.
Septic and toxic reactions may be life threatening. If bacterial contamination is
suspected, the transfusion must be stopped immediately and institutional guidelines
for investigating a reaction strictly followed. Usual investigation will include urgent
patient and blood unit culture and Gram stain. Initial treatment involves managing the
haemodynamic complications of sepsis and administration of intravenous antibiotics
covering the usual pathogens associated with transfusion-related sepsis.
A number of measures are used by NZBS to minimise the risk of bacterial contamination
in blood components. These include:
Page 146
Transfusion Medicine Handbook 3rd Edition
■
Predonation identification and deferral of potentially bacteraemic donors
■
Enhanced disinfection of the skin at the phlebotomy site
■
Diversion of the initial 10 - 40 mL blood collected into a separate container
■
Bacterial monitoring of platelet components using an automated bacterial
detection system
Visual inspection of the blood component for abnormal appearance (such as
discoloration or haemolysis) should be carried out both prior to release from the
Blood Bank and before administration. Blood components must not be transfused
beyond their expiry date.
7.10 Post-transfusion Purpura
Post-transfusion purpura (PTP) is a rare but potentially lethal complication of
transfusion of red cells and platelets. It is characterised by the sudden onset of severe
thrombocytopenia, typically 5 - 12 days following transfusion, often associated with
haemorrhage. PTP is most often seen in females (90% of cases) and in particular
those with a history of pregnancy.
Transfusion causes an anamnestic response boosting human platelet-specific antigen
(HPA) antibodies previously stimulated by pregnancy or earlier transfusion. The resulting
thrombocytopenia is due to alloantibody-mediated destruction of the transfused
platelets as well as “bystander” destruction of the patient’s own platelets.
Treatment of choice is high dose intravenous immunoglobulin, 2 g/kg body weight,
administered in divided doses over 2 - 5 consecutive days.
Plasma exchange and corticosteroids have been used in the past but an increase in
platelet count is significantly delayed when compared to intravenous immunoglobulin.
If platelet transfusion is unavoidable, platelets that are compatible with the patient’s
antibody should be used although survival of the platelets may be impaired during the
acute phase of the syndrome. If future transfusions are planned, red cell or platelet
components from donors negative for the implicated HPA antigen(s) should be selected
wherever possible.
Expert advice from a NZBS Transfusion Medicine Specialist or specialist haematologist
is needed when managing PTP.
7.11 Transfusion-Associated Circulatory Overload
When too much fluid is transfused or the transfusion is too rapid for a patient, fluid
overload can lead to systemic and pulmonary venous engorgement. Cardiogenic
pulmonary oedema and acute respiratory failure may follow.
The features of transfusion-associated circulatory overload (TACO) include acute
respiratory distress, tachycardia, increased blood pressure, evidence of fluid overload,
an enlarged cardiac silhouette and new or worsening pulmonary oedema in the chest
xray. TACO usually occurs within 6 hours of completion of the transfusion. Evidence
of fluid overload may include a documented positive fluid balance and/or a clinical
response to diuretic therapy. Diagnosis is supported by an elevated serum B-type
natriuretic peptide (BNP) or the accompanying N-terminal fragment (NT-pro BNP) to
Transfusion Medicine Handbook 3rd Edition
Page 147
more than 1.5 times the pretransfusion value (if available) and/or an increase in mean
arterial pressure or increase pulmonary wedge pressure.
Standard medical treatment includes stopping the transfusion, sitting the patient
upright, administering oxygen and diuretic therapy. Where necessary, vasodilator
therapy and/or non-invasive ventilatory support with continuous positive airways
pressure (CPAP) may be helpful. Venesection can also be considered.
TACO is most commonly seen in patients with low body weight, the elderly, infants
or children, those with a history of cardiac, respiratory or renal insufficiency, and in
the setting of red cell transfusion for chronic anaemia. Volume overload is a special
risk with albumin solutions.
Patients with chronic anaemia are normovolaemic or hypervolaemic and may have
signs of cardiac failure before any fluid is infused. Each unit should be given slowly and
the patient closely observed. Preemptive diuretic therapy may be helpful.
7.12 Transfusion-Related Acute Lung Injury
Transfusion-related acute lung injury (TRALI) is a significant transfusion-related
event. Although poorly recognised and undoubtedly underreported, international
haemovigilance data indicates that it is one of the most common causes of fatal
transfusion reactions.
TRALI is characterised by acute respiratory distress due to non-cardiogenic
pulmonary oedema, developing during or within 6 hours of transfusion. Typically,
plasma components containing antibodies against the patient’s white blood cells are
implicated. Transfusion is followed by a (usually) severe reaction with acute respiratory
distress, accompanied by chills and/or fever. The chest xray shows numerous, mainly
perihilar, nodules with infiltration of the lower lung fields without cardiac enlargement
or engorgement of the vessels. A transient leucopenia or neutropenia may be seen.
The implicated donors are almost always al oimmunised multiparous women. However
the number of reactions seen where antibodies are either not identified or serologically
cannot be implicated suggests the involvement of other mechanisms and risk factors.
The diagnosis of TRALI is therefore a clinical and radiographic diagnosis and is not
dependent on the results of laboratory tests or any proposed pathophysiologic
mechanisms. TRALI should be considered a clinical syndrome rather than a disease
with single cause. Treatment usually involves intensive care respiratory support.
Reporting to the NZBS Transfusion Medicine Specialist is essential so that an implicated
donor can be removed from the panel.
Testing of donors implicated in TRALI events
One proposed mechanism for TRALI is the interaction between human leucocyte
antigen (HLA) or neutrophil-specific (HNA) antibodies of donor origin and the recipient’s
white cells. NZBS has developed a standard procedure for investigating TRALI events,
including relevant serological testing. The investigation includes testing of the donor(s)
and recipient for HLA class I and II antibodies (identifying specificity if detected) and
for HNA antibodies. Antibody detection and identification is complemented by HLA
typing to confirm presence of the corresponding antigen(s). A crossmatch between
donor serum and recipient white cells is also useful, with a positive result strongly
implicating the particular donor(s).
Page 148
Transfusion Medicine Handbook 3rd Edition
TRALI risk reduction
A number of countries, including New Zealand, have introduced a strategy for reducing
the frequency of TRALI involving the use of FFP manufactured from plasma collected
only from male donors. The use of male-only donors for FFP appears to reduce
the incidence of TRALI. In addition, female plateletpheresis donors with a history of
pregnancy to > 20 weeks gestation are tested for the presence of HLA antibodies.
Where positive, donors are deferred from donating apheresis platelets and whole
blood donations are excluded from processing to platelet pools.
The differential diagnosis of TACO and TRALI
Acute respiratory distress during or shortly following transfusion may be due to TACO,
TRALI, a severe allergic reaction or the patient’s underlying condition. Unfortunately,
many of the early signs and symptoms are not discriminatory and can occur in other
types of transfusion reactions. Most FNHTR and allergic reactions can however be
readily identified as such.
It is important to distinguish between TACO and TRALI because of the relatively high
mortality for TRALI. Invasive measurements such as central venous and pulmonary
wedge pressures may be useful (elevated in TACO) but are not consistently diagnostic
or readily available, particularly in less severe cases. It has been suggested that
measurement of serum B-type natriuretic peptide (BNP) or the accompanying
N-terminal fragment (NT-pro BNP) might be useful in the differential diagnosis of TACO.
BNP is secreted from the cardiac ventricles as a result of ventricular pressure overload
and volume expansion, such as occurs with TACO. Low levels of BNP can help exclude
TACO however, whilst high levels may favour TACO they do not necessarily exclude
TRALI or allergic reactions, as these can co-exist.
7.13 Transfusion-Associated Dyspnoea
Only a minority of transfusion reactions are associated with predominantly respiratory
features however these are some of the most serious transfusion-related adverse
events. Included in this group are TACO, TRALI and allergic reactions, particularly of
the more severe type.
The term transfusion-associated dyspnoea (TAD) is used by haemovigilance
programmes to record events with significant respiratory distress, occurring within 24
hours of transfusion, that do not meet the criteria of TRALI, TACO or allergic reaction
nor are explained by the patient’s underlying condition.
7.14 Transfusion-Associated Graft-Versus-Host Disease
Transfusion-associated graft-versus-host disease (TA-GVHD) is a rare complication of
transfusion caused by engraftment and proliferation of transfused donor T-lymphocytes
which destroy recipient cells carrying “foreign” human leucocyte antigens (HLA).
Immunodeficient patients such as allogeneic bone marrow transplant recipients
receiving cellular components and fetuses receiving intrauterine transfusions are at
special risk for this disease. TA-GVHD has also occurred in immunologically normal
patients after transfusion of blood from a relative.
TA-GVHD is fatal in almost all cases. Acute TA-GVHD begins from 4 - 40 days after
transfusion with high fever followed by a diffuse erythematous skin rash progressing
to erythroderma and desquamation. Gastrointestinal and liver dysfunction occur and,
unlike GVHD following stem cell transplants, pancytopenia is common.
Transfusion Medicine Handbook 3rd Edition
Page 149
TA-GVHD is prevented by gamma irradiation of cellular blood components (red cells
and platelets) to a minimum dose of 25 Gray (Gy) targeted to the central position of
the container and 15 Gy to all other parts of the container. The irradiation dose must
not exceed 50 Gy.
For further information regarding irradiation of blood components see Section 4.6:
Irradiation.
7.15 Iron Overload / Haemosiderosis
Transfusion-dependent patients receiving red cells over a long period become
overloaded with iron. Chelation therapy may be used to minimise or reverse
accumulation of iron for these patients.
7.16 Transfusion-Related Immunosuppression
Allogeneic blood transfusion has been shown to cause suppression of the recipient’s
immune system. However, the mechanism behind the effect and the consequences
resulting from such transfusion-related immunomodulation (TRIM) remain unclear,
with contradictory evidence provided by both individual studies and metanalyses
performed to date.
Evidence from a number of studies suggests that allogeneic blood transfusion
enhances the survival of renal allografts, increases the recurrence rate of resected
malignancies, increases the incidence of postoperative bacterial infections and
increases the postoperative mortality rate from causes other than postoperative
infection.
7.17 Transfusion-Transmitted Infection
The perception of the risks of transfusion have been greatly influenced by HIV
transmissions that occurred before today’s safer testing procedures were available.
Blood donors, like anyone else, can occasionally carry an infectious agent, sometimes
for a long period, without having any clinical signs or symptoms. For this reason
donors are interviewed at each and every visit and laboratory tests are performed on
every blood donation. No part of the donation can be released until all these tests are
known to be clear. Computer blood management systems (BMS) are used to ensure
that this process is strictly adhered to.
There is very good evidence that with the donor selection and testing procedures used
by NZBS, the risk in New Zealand of infection through the contamination of blood
components and fractionated products is extremely small.
Table 7.2: Estimated Residual Risk of Transfusion-Transmitted Infection in New Zealand
Agent
Mean Window
Residual Risk (with 95% prediction
Period (days)
intervals)
HIV
5.6
1 in 9.2 million donations (2.5 - 33 million)
HCV
3.1
1 in 6.9 million donations (3.6 - 12 million)
HBV1
23.9
1 in 0.8 million donations (0.4 - 1.4 million)
1 HBV residual risk does not take into account the risk from occult HBV nor the proportion of recipients who
are HBV-immune from vaccination or past infection.
Page 150
Transfusion Medicine Handbook 3rd Edition
7.18 Other Infectious Agents
There are a number of other viral and parasitic infections that can be transmitted by
transfusion, for example malaria (
Plasmodium species), Chagas disease (
Trypanosoma
cruzi), Dengue, Zika and West Nile viruses. These are not endemic in New Zealand
but a blood donor may be exposed or become infected during travel or residence
abroad. The risk of acquiring Creutzfeldt-Jakob disease (CJD) and variant CJD from
blood transfusion remains very low and neither has ever been reported in New Zealand.
Rare cases have been reported in the UK.
It is important for NZBS to identify at-risk donors who will then either be selectively
deferred from donating or tested for evidence of infection.
7.19 Adverse Event Data
The NZBS National Haemovigilance Programme has been collecting data regarding
transfusion-related adverse events (TRAE) since 2005. The imputability scores of
adverse events have been recorded since 2008. From January 2008 to December
2013, a total of 2756 events having an imputability score ≥3 were reported for
transfusions involving 207,361 recipients who between them received 918,918 red
cell, platelet or frozen plasma components. The following table outlines the type and
frequency of TRAE during the period 2008 - 2013.
Table 7.3: Frequency of Transfusion-Related Adverse Events (Imputability1 ≥
3) Reported
to New Zealand Haemovigilance 2008 - 2013
Per 100,000
Per 10,000
Event
Number
Components
Recipients
Transfused
Transfused
FNHTR
1152
125.4
55.6
Allergic
920
100.1
44.4
UCT
179
19.5
8.6
TACO
119
13.0
5.7
IBCT
84
9.1
4.1
TAD
76
8.3
3.7
DSTR
60
6.5
2.9
Hypotension
60
6.5
2.9
DHTR
36
3.9
1.7
Inappropriate
transfusion
26
2.8
1.3
TRALI
13
1.4
0.6
Acute reaction2
12
1.3
0.6
Pain
10
1.1
0.5
TTI
9
1.0
0.4
All Events
2,756
299.9
133.0
1 Imputability ≥
3: the event is possibly, probably or certainly attributable to transfusion.
2Includes acute haemolytic and other severe acute transfusion reactions.
Transfusion Medicine Handbook 3rd Edition
Page 151
Key
FNHTR
Febrile non-haemolytic transfusion reaction
Allergic
Allergic transfusion reaction
TACO
Transfusion-associated circulatory overload
IBCT
Incorrect blood component transfused
UCT
Unclassifiable complication of transfusion
TAD
Transfusion-associated dyspnoea
DSTR
Delayed serologic transfusion reaction
DHTR
Delayed haemolytic transfusion reaction
TRALI
Transfusion-related acute lung injury
TTI
Transfusion-transmitted infection
7.20 Other Complications
Complications associated with large volume transfusions including hypocalcaemia,
hyperkalaemia, hypothermia, disturbances of acid base balance and adult respiratory
distress syndrome are considered in Section 6.3:
Complications of Acute Blood Loss
Associated with Large Volume Transfusions.
Page 152
Transfusion Medicine Handbook 3rd Edition
8
CLINICAL ALTERNATIVES AND APPLICATIONS
There are no effective alternatives to red cells, platelets, white cells, haemopoietic
progenitor cells, plasma and most fractionated products.
In this section, the emphasis, with some exceptions, is directed towards minimising
exposure to allogeneic blood components. One such option is the collection and
transfusion of autologous blood components, namely red cells, platelets, fresh or frozen
plasma and some blood constituents such as serum eye drops. Practices which include
preoperative collection, acute normovolaemic haemodilution (ANH) and perioperative
salvage are all essentially modifications of routine blood transfusion practice. However,
the circumstances where these procedures are indicated or applicable are limited.
8.1
Autologous Blood Collection and Transfusion
Using autologous blood will avoid the risks of alloimmune complications of transfusion and
may reduce the risk of many transfusion-transmitted infections. Autologous transfusion
does not however reduce the risk of bacterial infection associated with transfusion.
Autologous blood can be collected several ways, including:
■
Preoperative collection where blood is collected from the patient during the
weeks leading up to their surgical procedure. This should only be performed
in exceptional clinical circumstances. In general, this is limited to patients
undergoing an elective procedure normally requiring a blood transfusion
(otherwise units of blood collected are likely to be wasted) where there are
clinical, religious or cultural reasons for preferring autologous blood. The
inevitable reduction in preoperative hemoglobin level increases the likelihood
that any intra- or post-operative blood transfusion (autologous or allogeneic)
may be required.
Autologous blood components obtained by preoperative donation are collected,
prepared and stored under the same conditions as allogeneic blood. Acceptance
of these individuals may vary from that of regular donors for allogeneic use,
particularly with reference to age. The preferred programme for obtaining up to
four units of blood involves collections at weekly intervals from approximately
four weeks before surgery, with the last unit collected no later than one week
before surgery. Most patients will require supplementary iron during the period
of autologous collections to maintain the haemoglobin > 110 g/L. The patient
will be required to pay for autologous collections unless this is undertaken for
clinical, religious or cultural reasons. The charge covers the costs of collection,
processing and testing of the units. Patients wishing to donate autologous blood
prior to surgery should initially discuss this with their surgeon who should in turn
liaise with a NZBS Transfusion Medicine Specialist.
■
Frozen autologous blood where red cells and platelets are collected from patients
with rare blood groups or multiple antibodies and then frozen for future use.
■
Acute normovolaemic haemodilution (ANH) where a patient donates whole
blood immediately before surgery, which is then replaced with intravenous colloid
or crystalloid solution to maintain normovolemia. The removed whole blood is
usually returned within several hours, for example at wound closure, providing
fresh clotting factors and platelets.
Transfusion Medicine Handbook 3rd Edition
Page 153
■
Red cell salvage
Intraoperative blood salvage: where blood is collected from the surgical site
using a cell-saver machine and reinfused after processing (washing and filtering)
during or after surgery.
Post-operative blood salvage: where blood is collected from wound drains and
reinfused. The salvaged blood may be processed before reinfusion to minimise
potential coagulation problems sometimes seen with unprocessed blood.
ANH and red cell salvage are usually performed under the supervision and the
responsibility of anaesthetists and/or surgeons.
8.2
Non-Blood Plasma Volume Expanders
The judicious use of plasma volume expanders can result in adequate restoration of
blood volume at an economic cost.
It should be noted however that positive fluid balance is an independent predictor
of poor outcome in ICU patients and this, together with the increasing adoption of
permissive hypotension (hypotensive resuscitation) in a number of clinical settings, is
leading to more restrictive intravenous fluid administration in the critically ill than has
been used in the past.
Crystalloid Solutions
Initial resuscitation should be with 1 - 3 litres of a crystalloid solution such as Plasmalyte
148, pH 7.4 (an isotonic non-calcium containing ‘balanced’ solution buffered with
acetate) or 0.9% sodium chloride (‘normal’ saline). Adequate crystalloid resuscitation
may often avert the need for other plasma expanders. Colloid products can then be
used in the recommended doses. If prolonged volume expansion is required, such as
in a severely ill trauma patient, then the use of albumin solutions rather than synthetic
colloids is likely to be justified.
Plasmalyte 148 in 5% glucose, dextrose solutions and compound sodium lactate (CSL)
solutions, e.g., Hartmann’s or Ringer-Lactate, must not be administered simultaneously
with a red cell infusion as the solutions are not compatible. No drugs or additives, other
than 0.9 % sodium chloride intravenous infusion, are recommended to be mixed with
red cells before or during transfusion.
Synthetic Colloid Solutions
Colloids should only be used if crystalloids are insufficient to stabilise a patient. Due
to the risk for acute renal damage and a requirement for dialysis, the use of synthetic
colloids, particularly starch solutions, should be avoided in critically ill patients and
those with sepsis or pre-existing renal dysfunction. In these situations, and where
colloids are required, the use of albumin solutions is likely to be justified.
Gelatin Solutions (Gelofusal®)
Gelofusal® is a 4% solution of succinylated gelatin prepared from heat degraded
cattle bone gelatin. It has an average molecular weight of 30,000 Dalton. Gelofusal®
has a volume-expansion effect that lasts three to four hours. The frequency of severe
acute reactions, which are usually anaphylactic, is < 1/10,000 and patients should be
closely monitored at least while the first 20 - 30 mL are infused. The maximum volume
Page 154
Transfusion Medicine Handbook 3rd Edition
that can be infused has not been determined but is at least 3 - 5 litres. Apart from a
dilutional effect on coagulation proteins, haemostasis is not affected. Gelofusal® does
not interfere with blood group analysis and cross-matching procedures.
Gelofusal® must not be administered simultaneously with a red cell infusion, as the
solution is not compatible.
Hydroxyethyl Starch (Voluven® 6%; Volulyte® 6%)
Hydroxyethyl starch (HES) solutions provide an alternative colloidal fluid to albumin
and plasma for use in plasma volume expansion. HES solutions are prepared from
chemically modified amylopectin.
Voluven® 6% and Volulyte® 6% are solutions of HES in 0.9% sodium chloride. The
average molecular weight is 110,000 - 150,000 Dalton. Infusion results in a plasma
volume increase of approximately 100% of the infused volume and the effect lasts four
to six hours. The frequency of severe acute reactions, which are usually anaphylactoid,
is 1/10,000 - 1/1000 and patients should be closely monitored at least while the first
10 - 20 mL are infused. With the administration of Voluven disturbances of blood
coagulation, beyond dilutional effects, can occur. The maximum volume recommended
before this product interferes with coagulation is about one litre per day.
8.3
Oxygen Carrying Compounds
Although red cell substitutes in the form of haemoglobin-based oxygen carriers,
including polymerised haemoglobin and haemoglobin conjugated with polyethylene
glycol, and perfluorocarbon oxygen carriers have been under investigation for many
years, there is no indication that these compounds will be widely available for routine
use in the near future.
8.4
Haemopoietic Growth Factors
Genetically engineered haemopoietic growth factors are expected to have an increasing
impact on the use of allogeneic blood components. These should only be used by
clinicians with relevant expertise.
Recombinant Human Erythropoietin (r-HuEPO)
■
Epoetin alfa - Eprex®, Binocrit®
■
Epoetin beta - NeoRecormon®
Administration of r-HuEPO can increase the rate of red cell production, principally
through its proliferative effect on erythroid precursors.
The use of r-HuEPO in patients with chronic renal failure has seen a significant reduction
in the requirement for repeated transfusion of red cells, allowing them to live virtually
transfusion-free and without symptomatic anemia. The benefit is seen prior to dialysis
and in dialysis-dependent patients. For patients awaiting renal transplantation, reducing
allogeneic red cell exposure may reduce the risk of adverse outcomes from immune
modulation and/or transfusional hemosiderosis.
Patients experiencing chronic anaemia with reduced levels of circulating erythropoietin
may require fewer red cell transfusions if treated with r-HuEPO. They include those with:
Transfusion Medicine Handbook 3rd Edition
Page 155
■
chronic inflammatory diseases such as rheumatoid arthritis
■
cancer, with or without chemotherapy treatment
■
myelodysplastic syndromes
There is evidence that the use of r-HuEPO along with iron supplements can reduce
transfusion requirements for anaemia of prematurity seen in infants with very low
birthweight (< 1000 g). However, studies have shown that the benefits are only seen
when treatment with r-HuEPO is initiated after two to four (or more) weeks of life.
Before this, the major factor affecting transfusion requirements for sick neonates is
iatrogenic blood loss, for which treatment with r-HuEPO is ineffective.
Eprex® and Binocrit® have the following indications:
■
Severe symptomatic anaemia of renal origin in patients with renal insufficiency
not yet undergoing dialysis.
■
Anaemia associated with chronic renal failure in paediatric and adult patients
on dialysis.
■
Chemotherapy-induced anaemia in patients with non-myeloid malignancies.
■
Adult patients with mild-to-moderate anaemia (haemoglobin 100 - 130 g/L)
scheduled for elective surgery with an expected moderate blood loss (i.e., 2 - 4
units or 900 - 1800 mL), to reduce exposure to allogeneic blood transfusion
and to facilitate erythropoietic recovery.
■
To augment autologous blood collection in anaemic adult patients undergoing
major surgery who are not expected to deposit preoperatively their complete
perioperative blood needs.
Granulocyte-Colony Stimulating Factor (G-CSF)
■
Filgrastim - Zarzio®, Neupogen®
■
Pegfilgrastim - Neulastim®
G-CSF is accepted therapy in the management of patients with severe neutropenia
associated with chemotherapy-induced bone marrow failure, autoimmune and drug-
induced disorders, congenital agranulocytosis and to increase the level of haemopoietic
progenitor cells (HPC) in the circulation prior to collection by apheresis for autologous
and allogeneic HPC transplantation.
CXCR4 Chemokine Receptor Antagonist
■
Plerixafor - Mozobil®
Plerixafor, an antagonist of the CXCR4 chemokine receptor, is used in combination
with G-CSF to enhance mobilisation of haematopoietic stem cells in adult patients
with myeloma and lymphoma whose cells mobilise poorly.
Thrombopoietin Receptor Agonist
■
Eltrombopag olamine - Revolade®
Eltrombopag binds to the thrombopoietin receptor (TPO-R) inducing proliferation and
differentiation of megakaryocytes with subsequent platelet production. Revolade® is
Page 156
Transfusion Medicine Handbook 3rd Edition
indicated for the treatment of adult patients with chronic immune thrombocytopenic
purpura (ITP) refractory to corticosteroids and immunoglobulins, and for the treatment
of thrombocytopenia in patients with chronic hepatitis C to allow the initiation and
maintenance of interferon-based therapy. Promising results following the use of
eltrombopag in other clinical areas including myelodysplasia, severe aplastic anaemia
refractory to immunosuppressive therapy, non-myeloablative chemotherapy and
thrombocytopenia due to HIV and liver disease are the subject of ongoing research.
8.5
Recombinant Coagulation Factors
These products should only be used by clinicians with relevant expertise.
Recombinant Factor VIII (rFVIII)
■
Moroctocog alfa - Xyntha®
■
Octocog alfa - Kogenate® FS, Advate, Recombinate™
rFVIII has similar efficacy to plasma-derived factor VIII in the management of bleeding
and a similar rate of development of inhibitors associated with deficiency of circulating
factor VIII (haemophilia A). It contains no von Willebrand factor (vWF) and therefore
should not be used for improving haemostasis in von Willebrand disease (vWD). Unlike
other rFVIII products, Recombinate™ contains human albumin, added to stabilise the
factor VIII and therefore could theoretically transmit blood-borne infections.
Recombinant Factor IX (rFIX)
■
Nonacog alfa - BeneFIX®
■
Nonacog gamma - Rixubis
■
Eftrenonacog alfa - Alprolix
rFIX has similar efficacy to plasma-derived factor IX in the management of bleeding
and a similar rate of development of inhibitors associated with deficiency of circulating
factor IX (haemophilia B, Christmas Disease). No human protein is added to stabilise
the product.
Recombinant Factor VIIa (rFVIIa)
■
Eptacog alfa - NovoSeven® RT
NovoSeven® RT is licensed for the treatment of bleeding episodes in patients with
haemophilia A or B who have inhibitors to factors VIII and IX respectively, in patients
with congenital factor VII deficiency and in patients with Glanzmann’s thrombasthenia
refractory to platelet transfusion due to GPIIb-IIIa and/or HLA antibodies.
8.6
Desmopressin acetate (Octostim®, Minirin®)
Desmopressin should only be used by clinicians with relevant expertise. It is a synthetic
analogue of the antidiuretic hormone vasopressin. When administered in high doses of
0.3 mcg/kg it releases factor VIII (FVIII) and von Willebrand factor (vWF) from endothelial
storage sites with a three to five-fold rise in plasma FVIII coagulant activity (FVIII:C) and
a three to four-fold rise in vWF level. Desmopressin has also been shown to lead to
improvement in, or normalisation of, assays of platelet function, including the bleeding
time, in patients with uremia, liver cirrhosis, congenital or substance-induced platelet
dysfunction. The effect of desmopressin on FVIII:C lasts for six to eight hours while
the effect on bleeding time is for a shorter period of one to three hours.
Transfusion Medicine Handbook 3rd Edition
Page 157
Indications for use
■
Mild to moderate Haemophilia A
■
von Willebrand disease (vWD)
- In most patients with type I vWD administration of desmopressin will increase
von Willebrand factor-antigen (vWF:Ag) level three to four-fold and shorten
or normalises the bleeding time.
- Some but not all type IIA vWD patients respond to desmopressin.
- In type IIB vWD administration of desmopressin is generally contraindicated
as it fails to shorten the bleeding time and may produce a severe transient
thrombocytopenia.
- Desmopressin is also contraindicated in pseudo von Willebrand’s disease.
- Type III vWD does not respond to desmopressin.
■
Congenital platelet function defects
■
Acquired platelet function defects
■
Uraemia
■
Hepatic cirrhosis
Contraindications
■
Habitual and psychogenic polydipsia
■
Unstable angina, myocardial infarction or stroke
■
Decompensated cardiac failure
■
Type IIB von Willebrand’s disease
Precautions
■
Hyponatraemia; with repeated doses, fluid restriction and monitoring of sodium
levels are recommended
■
Patients at risk for raised intracranial pressure, including closed head injury
■
Caution when using in children under 2 years of age
■
Caution when using in patients > 70 years of age, especially those with a history
of vascular disease
Presentation
The product is presented as 1 mL ampoules containing 15 mcg (Octostim®) or 4
mcg (Minirin®) of desmopressin acetate. Administration is either by intravenous or
subcutaneous injection. A suitable intranasal preparation is not currently available in
New Zealand.
Page 158
Transfusion Medicine Handbook 3rd Edition
Dosage and administration
Desmopressin acetate 0.3 mcg/kg for intravenous administration can be diluted in
50 - 100 mL of isotonic saline and given no more than one hour before surgery. The
first 5 mL is given over five minutes and, provided the patient does not show marked
tachycardia or other adverse reactions, the remainder of the dose may be given over
the next 15 - 30 minutes. When administered subcutaneously, Octostim® should be
given one hour preoperatively.
If a positive effect is obtained, the initial dose may, if necessary, be repeated once after
24 hours, with sodium monitoring and fluid restriction, and no more than two doses in
any five day period. Octostim® given subcutaneously and once daily is the preferred
method to achieve adequate release of FVIII and vWF while minimising side effects.
Patients may become progressively unresponsive to desmopressin with repeated
daily doses over two to three days. Responsiveness will return when the drug has
been discontinued for two days. For this reason it is not recommended to perform a
desmopressin trial within four days of planned surgery.
8.7
Tranexamic Acid (Cyklokapron®)
Tranexamic acid is an inhibitor of fibrinolysis. Initially this compound was used primarily
to reduce mucosal bleeding in patients with haemophilia and von Willebrand disease.
Currently it is also used to decrease blood loss in cardiopulmonary bypass and joint
surgery, trauma-related bleeding, obstetric complications, severe hypoproliferative
thrombocytopenia, disorders of platelet dysfunction (e.g., Glanzmann’s thrombasthenia),
following treatment with anti-platelet medications and in conditions associated with
increased fibrinolysis including menorrhagia, prostatectomy, cervical conisation,
gastrointestinal bleeding, dental extraction and epistaxis. Additionally, tranexamic acid
has a role in the management of hereditary angioedema.
Use of tranexamic acid carries a risk of clot formation resistant to fibrinolytic therapy
and is therefore generally contraindicated in thromboembolic disease and in DIC. Due
to the risk of clot-induced hydronephrosis, tranexamic acid is contraindicated with
haematuria/bleeding from renal parenchyma.
Dose adjustment is required with even mild renal impairment.
Presentation
Cyklokapron® is presented as 500mg oral tablets or 5 mL ampoules containing 500
mg tranexamic acid. Administration of the latter is either by IV injection or, in the case
of epistaxis or dental surgery, topical via application of soaked gauze.
Dosage and administration
The recommended standard dose of tranexamic acid is 1000 - 1500 mg orally or
500 - 1000 mg IV at a rate of 1 mL/minute, two to three times per day. In general,
the use of tranexamic acid intravenously is recommended only when adequate doses
cannot be administered orally.
In the setting of surgery, tranexamic acid 10 - 15 mg/kg administered IV over 10
minutes may be given as preoperative prophylaxis or as therapy for bleeding. Further
intravenous infusion or bolus IV doses of 500 - 1000 mg, every 8 hours, may be given.
Transfusion Medicine Handbook 3rd Edition
Page 159
Oral doses up to 1500 mg, administered three to four times daily, may be used for up
to 14 days to cover the post-operative period.
For trauma-related bleeding, tranexamic acid should be initiated within 3 hours of
the event.
For oral mucosal bleeding, experience suggests clinical effect can be achieved from
the use of a crude mouth wash made by dispersing 500 mg tranexamic acid tablet
to a slurry in 10 - 15 mL water and swirling the total preparation around the mouth
for two minutes before expelling. This is used four times daily for up to seven days.
8.8
Iron Supplementation
Preoperative identification and treatment of iron deficiency anaemia may avoid pre- and/
or post-operative red cell transfusion in elective surgery. Correction of iron deficiency,
even in the absence of anaemia, may improve erythropoietic recovery and reduce
the risk for red cell transfusion in post-operative patients. Where red cell transfusion
is required (e.g., for cardiac compromise), iron therapy should always follow, as
transfusion fails to replenish deficient iron stores.
The following algorithm is adapted from the Australian National Blood Authority (NBA)
Preoperative Haemoglobin Assessment and Optimisation Template of the
Perioperative
(2012) Patient Blood Management Guidelines.
Page 160
Transfusion Medicine Handbook 3rd Edition

Transfusion Medicine Handbook 3rd Edition
Page 161
9
OTHER SERVICES PROVIDED BY NZBS
9.1
Therapeutic Apheresis
Some NZBS centres provide therapeutic apheresis programmes using a centrifugal
cell separator. Procedures may be carried out in a hospital ward or, if there is a medical
officer on site and patients are mobile and in good health, at a NZBS blood centre.
Therapeutic plasma exchange (TPE) is an established first-line treatment in some
conditions and second-line treatment in several others. TPE may be combined
with other medical therapy in managing diverse conditions such as hyperviscosity
syndromes, Guillain-Barré syndrome, thrombotic thrombocytopenic purpura, chronic
inflammatory demyelinating polyneuropathy, familial hypercholesterolaemia and renal
transplant rejection. It is also sometimes used for the treatment of myasthenia gravis,
polymyositis, SLE and other autoimmune disorders. The potential hazards of plasma
exchange should be taken into account when considering this treatment.
Leucopheresis may be used to reduce leucostasis in patients with very high white
cell counts. Similarly, plateletpheresis may occasionally be used for patients with
complications due to thrombocytosis.
Replacement fluids used for TPE usually include albumin or fresh frozen plasma (FFP).
Saline, usually in combination with albumin, can also be used in certain conditions
such as hyperleucocytosis and thrombocytosis. FFP is occasionally used to correct a
deficiency of coagulation factors and cryosupernatant may be used for patients with
thrombotic thrombocytopenic purpura.
Complications of therapeutic apheresis include allergic reactions to FFP or
cryosupernatant, volume overload or hypovolaemia, air embolism, haemolysis,
extracorporeal clotting, citrate toxicity, coagulopathy and vasovagal attacks. However,
present procedural methods appear to minimise these complications.
To request therapeutic apheresis, contact a NZBS Transfusion Medicine Specialist/
Medical Officer.
9.2
Therapeutic Venesection
Whole blood therapeutic venesection is available for patients with medical conditions
where regular venesection is beneficial such as haemochromatosis, polycythaemia
and porphyria cutanea tarda.
Blood collected from an individual with genetic haemochromatosis may potentially
be used to prepare therapeutic blood components and fractionated products if the
patient meets all normal donor selection criteria and is registered as a blood donor.
See Section 2.7:
Haemochromatosis for further information.
In the case of polycythaemia vera managed by venesection in combination with
cytoreductive medication, the referring doctor remains responsible for the management
of the patient’s underlying condition and should regularly review the patient (at least
every 12 weeks), as well as providing instructions concerning the frequency of
venesection. NZBS is responsible for the venesection collection and for ensuring the
safety of the patient during the procedure. For patients with polycythaemia managed
by venesection alone, and following clear instruction from the referring doctor, it may be
Page 162
Transfusion Medicine Handbook 3rd Edition
possible for NZBS to monitor the haematocrit and adjust the frequency of venesection
accordingly. In such cases, review by the referring doctor at intervals greater than 12
weeks may be appropriate.
A similar arrangement for the monitoring of ferritin levels and titration of therapeutic
venesection may be made for patients with porphyria cutanea tarda.
9.3
Tissue Bank
The NZBS Tissue Banks provide:
■
Cadaver split skin, sourced locally and from international tissue banks, used as
a physiological dressing to treat victims of burns.
■
Donated or autologous bone, mostly femoral heads, used in reconstructive
orthopaedic surgery.
■
Autologous cranial bone flaps held for neurosurgical patients until reimplantation
weeks to months later.
NZBS Tissue Banks are located in the NZBS centres in Auckland, Hamilton, Palmerston
North, Wellington, Christchurch and Dunedin. Cadaver skin is only available from the
Auckland centre. Donated bone is held at the NZBS sites as well as a number of
DHB-managed blood banks.
All donations are voluntary. Written consent is required from the donor or, in the case
of skin, from next of kin. All donations are tested for infectious diseases and bacterial
contamination. Frozen tissue is generally stored frozen and held for up to five years.
All tissue is issued frozen to a named patient and is only for that patient. An information
sheet is available from Tissue Bank for consenting patients. Unused frozen tissue
should be returned to the Tissue Bank. Unused thawed tissue must be discarded.
9.4
Autologous Serum Eye Drops
Autologous serum eye drops (SED), a blood component prepared by NZBS, is used
for treating dry eyes and corneal epithelial defects. It is not normally first-line treatment
due to the cost and requirement that patients donate blood. However, in selected
patients, SED have proven to be a successful treatment modality.
Conditions for which SED can be used include severe Sjögren’s syndrome, Stevens-
Johnson syndrome, ocular cicatricial pemphigoid, some causes of epithelial limbal
stem cell deficiency such as chemical or thermal ocular surface burns and other
severe ocular surface disorders that do not respond to conventional therapies. SED
may also be useful in acute management of severe sight threatening ocular surface
chemical or thermal burns.
Although not fully defined, it is believed that epidermal growth factor, transforming
growth factor ß, vitamin A, platelet-derived growth factor and fibronectin are important
serum constituents that promote healing of the ocular surface.
SED is prepared from a single blood donation and contains no added preservatives
or antimicrobial agents. It has the potential to transmit infectious diseases or become
contaminated with microorganisms. Accordingly, blood collected for SED must meet
Transfusion Medicine Handbook 3rd Edition
Page 163
standard safety requirements. This includes a donor health assessment and screening
of the blood donation to exclude evidence of human immunodeficiency virus, hepatitis
B virus and hepatitis C virus. Following manufacture of SED, a proportion of the eye
drop bottles are cultured for 14 days to confirm bacterial sterility.
To access the service, ophthalmologists complete a specific request form that is
forwarded to NZBS. As this constitutes a prescription, it is valid for three months
only, as per the Medicines Act, although blood is only collected every six months.
Due to the time required for manufacturing and sterility testing, 18 working days are
normally required from donation to issuing of SED. In exceptional circumstances and
with the patient’s consent, the ophthalmologist may request that SED are issued prior
to completion of the sterility testing.
Appropriate storage of SED vials is essential to minimise the risk of bacterial
contamination. Vials are stored frozen in a NZBS facility or hospital blood bank and
are dispensed in quantities necessary to cover a 4 week period. The dispensed vials
should be kept frozen until needed, usually in the patient’s domestic freezer. Each
vial usually lasts one week and should be stored in a refrigerator once thawed. Any
adverse events should be reported to NZBS for investigation.
For patients where blood cannot safely be donated or where allogeneic SED are the
preferred product, for example in ligneous conjunctivitis, NZBS can, on a named
patient basis, prepare SED from an allogeneic donation. NZBS does not hold this
as a stock item.
9.5
Reference Laboratory (Immunohaematology)
The NZBS Reference Laboratory is located in the NZBS Auckland centre and offers a
national immunohaematology testing service as well as clinical and technical advice for
resolving problems encountered during pretransfusion and antenatal testing such as:
■
blood grouping, antibody screening and crossmatching
■
antibody identification/confirmation and resolution of difficult antibody mixtures
■
antibody titration (antenatal, cold agglutinins and isoagglutinins)
■
adsorption and elution studies for patients with autoantibodies
■
red cell phenotyping and genotyping
9.6
Tissue Typing Laboratory
The NZBS Tissue Typing Laboratory is also located in the NZBS Auckland centre and
is responsible for tissue typing tests in support of New Zealand’s bone marrow and
solid organ transplant programmes. The laboratory also carries out testing for disease
association markers, for antibodies implicated in transfusion-related reactions, for
antibodies against platelet antigens and is responsible for the provision of compatible
or matched platelets for transfusion.
The following provides an indication of the wide range of white cell and platelet-related
investigations undertaken:
■
HLA Class I and Class II typing
Page 164
Transfusion Medicine Handbook 3rd Edition
■
HLA-antibody screening
■
T- and B-lymphocyte crossmatching
■
platelet-associated antibody testing
■
serum platelet antibody testing
■
platelet crossmatching
■
fetal and neonatal alloimmune thrombocytopenia (FNAIT) investigation
■
platelet genotyping
Transfusion Medicine Handbook 3rd Edition
Page 165
10
NZBS SAMPLE REQUIREMENTS
It is not possible to overemphasise the importance of proper patient identification.
Most errors relating to transfusion practice arise from administrative and clerical error.
These errors can have serious consequences for patients and are sometimes fatal.
Collection and labelling of blood samples for pretransfusion testing is described in
Section 3.6:
Collecting Blood Samples for Pretransfusion Testing.
The sample acceptance criteria used by NZBS are based on the 2016 Australian &
New Zealand Society of Blood Transfusion (ANZSBT)
Guidelines for Transfusion, Pre
and Postnatal Immunohaematology Testing. Blood Bank staff are not authorised to
accept samples which do not meet labelling requirements. Where necessary these
will be rejected and new samples requested. Where a dispute arises in relation to a
sample, the final decision on suitability for testing will lie with an NZBS Transfusion
Medicine Specialist/Medical Officer.
The following tables provide a brief guide to the types of samples NZBS requires
for laboratory investigations. Specific guidance can be obtained from the individual
laboratories and NZBS Transfusion Medicine or Nurse Specialists.
Page 166
Transfusion Medicine Handbook 3rd Edition
op
Tube T
Pink
Pink
Pink
Pink
Pink
N/A
Pink
om transfused unit(s)
ed
e-transfusion sample
2 A
A
A
A pr
A post-transfusion sample
A
emnants fr
Sample Requir
1 x 6 mL EDT
1 x 6 mL EDT
1 x 6 mL EDT
1 x 6 mL EDT
1 x 6 mL EDT
Giving set/r
2 x 6 mL EDT
e
oup &
equir
ovision
etransfusion
ovision of blood
, not fractionated
1
een in pr
esting
e antibody identification
equir
een’
oducts
esence of autoantibodies may r
Comment
Normally performed as part of a ‘Gr
Scr
A positive antibody scr
testing will r
which may delay the pr
Reactions observed with transfusion of
blood components only
pr
Pr
adsorption techniques to detect underlying
alloantibodies which may delay pr
of blood
ransfusion Medicine Specialist for specific sample requirements or if further information is required.
oup
eaction investigation
Samples Required for Pretransfusion T
een
oup & scr
Table 10.1:
Test
ABO / RhD gr
Gr
Red cell antibody identification
Transfusion r
Autoimmune haemolytic anaemia
Contact the Blood Bank or T
Ethylenediaminetetraacetic acid.
1
2
Transfusion Medicine Handbook 3rd Edition
Page 167
op
Tube T
Pink
Pink
Pink or lavender
Pink or lavender
Pink
A
EDT2
A
ed
A
A
A
Sample Requir
1 x 6 mL EDT
1 x 6 mL EDT
1 x 6 mL or 4 mL EDT
1 x 6 mL or 4 mL or 0.5 mL
1 x 6 mL EDT
1
nal blood contamination
esting
.
Comment
Tested for mater
Maintain and transport samples at 37°C
T)
ransfusion Medicine Specialist for specific sample requirements or if further information is required.
d blood
een and titration
eening
T on cor
oup & DA
ect antiglobulin test (DA
Table 10.2: Samples Required for Diagnostic T
Test
Antenatal scr
Antenatal antibody titration
Gr
sample
Dir
Cold agglutinin scr
Contact the Blood Bank or T
0.5 mL pink top microcontainer for use in infants only
1
2
Page 168
Transfusion Medicine Handbook 3rd Edition
op
Tube T
Pink
Pink
Pink
Pink
Pink
Pink
N/A
Pink
Pink
Pink or lavender
Pink
om transfused unit(s)
A
ed
e-transfusion sample
A
A
A
A
A pr
A post-transfusion sample
A
A
A
emnants fr
Sample Requir
2 x 6 mL EDT
1 x 6 mL EDT
1 x 6 mL EDT
2 x 6 mL EDT
1 x 6 mL EDT
1 x 6 mL EDT
Giving set/r
1 x 6 mL EDT
1 x 6 mL EDT
1 x 6 mL or 4 mL EDT
1 x 6 mL EDT
1
esting
y Diagnostic T
d blood sample
egnancies beyond 16 weeks
Comment
Pr
Cor
ransfusion Medicine Specialist for specific sample requirements or if further information is required.
nal
4
y or T
oblem
T
om mater
ransfusion Medicine Specialist required
ouping pr
eaction investigation
5
n
with positive DA2
3
nal phenotyping: HDFN
Table 10.3: Samples Required for Reference Laborator
Test
Alloantibody investigation
ABO / Rh gr
Extended RBC phenotype
AIHA
Transfusion r
Compatibility testing
Fetal D genotyping fr
blood
Haemolytic disease of the
newbor
Pater
and FNAIT
Contact the Reference Laborator
Autoimmune haemolytic anaemia.
Prior discussion with T
Haemolytic disease of the fetus and newborn.
Fetal and neonatal alloimmune thrombocytopaenia.
1
2
3
4
5
Transfusion Medicine Handbook 3rd Edition
Page 169
op
Tube T
Yellow
Red
Pink
Yellow
Red
Red
Yellow
Red
Pink
Yellow
Red
Yellow
Yellow
Red
Red
Yellow
A tubes.
2
ed
3
A
A
Sample Requir
4 x 10 mL CPDA
1 x 10 mL clotted
1 x 6 mL EDT
1 x 10mL CPDA
1 x 10mL clotted
1 x 10mL clotted
4 x 10 mL CPDA
1 x 10 mL clotted
1 x 6 mL EDT
4 x 10 mL CPDA
1 x 10 mL clotted
1 x 10mL CPDA
4 x 10 mL CPDA
2 x 10 mL clotted
2 x 10 mL clotted
2 x 10 mL CPDA
A samples may alternatively be sent in 7 mL lavender EDT
enal transplant
1 y
ogenitor cell (HPC) or solid
esis platelet donor
yping Laborator
colepsy
issue T
Comment
Haematopoietic pr organ transplant patient or potential donor and apher
e.g., ankylosing spondylitis, coeliac disease, nar
HLA antibodies
Patients awaiting cadaveric r
Solid organ transplant patient or potential donor
Platelet and platelet-associated antibody
Donor
Patient
ransfusion Medicine Specialist for specific sample requirements or if further information is required.
y or T
een /
A) genotyping
een
yping Laborator
ossmatch
issue T
efractory to platelets
ossmatch
Table 10.4: Samples Required for T
Test
HLA typing
(initial or confirmatory)
Disease association
Monthly serum tray
HLA antibody scr
Lymphocyte cr
Platelet antibody scr cr
Platelet antigen (HP
Patients r
TRALI investigation
Contact the T
Clotted samples may alternatively be sent in 3 x 5 mL or 2 x 7 mL red top tubes. EDT
Citrate phosphate dextrose adenine.
1
2
3
Page 170
Transfusion Medicine Handbook 3rd Edition
Prepared by the New Zealand Blood Service.
Private Bag 92071, 71 Great South Road, Epsom, Auckland 1142
Telephone 09 523 5733 Fax 09 523 5754
www.nzblood.co.nz
111G12203 06/16
NZBCL159